Lacking MB C Class I Gene Products * |
|
* This work has been supported by U .S. Public
Health Service Grants 5 ROI CA25250-06 and 1 ROI CA-44882-01 awarded
by the National Cancer Institute, The Swedish Cancer Society, The
Swedish Society for Medicine, the Bristol Myers Company, and the
Royal Swedish Academy of Sciences.
Natural killer (NK) cells represent an important subset of lymphocytes. These cells fulfil crucial cytotoxic as well as regulatory functions of the immune system. Compromised NK cell activity has been found to be associated with the development of several diseases, including cancer, AIDS and virus infections. Intact NK cell activity appears to play an important role in health. More recent data suggests that NK cells may be involved in the pathogenisis of some human diseases and serve as an early predictor for susceptibility to disease [1]. In transplantation, NK cells participate in rejection of allogeneic bone marrow cells [2]. The molecular basis for the ability of NK cells to discriminate between normal and aberrant cells is not known in detail [3-5]. The aim of the present overview is to discuss studies of how target cell major histocompatibility complex (MHC) class I expression affects NK-target interactions and ultimately target susceptibility to NK cell mediated lysis. The rationale for current studies is a hypothesis which has provided one test able model for self- non-self discrimination by NK cells [4, 5]. This hypothesis was originally based on (a) the patterns common to the various cytotoxic reactions attributed to NK cells and (b) a comparative analysis of the different strategies employed by vertebrates and invertebrates in order to distinguish between self and non-self. This hypothesis, presented as the "missing self" hypothesis [3], provided testable predictions for the investigation of the influence of the MHC class I gene products by NK cells. Briefly, it was suggested that NK cells could kill certain target cells because the latter express reduced levels of MHC class I molecules. This model underlies the work presented below. However, before going into the missing self hypothesis, present knowledge of NK cells and in particular their cytotoxic activity is briefly recapitulated.
The ability to kill certain tumour cell lines in vitro, without prior immunization or sensitization, was the first attribute of NK cells to be identified (reviewed in [6]). While this reaction was investigated in detail in the late 1970s several new insights as to their morphology, cell surface marker expression, tissue expression and action have become evident during the last 10 years (for reviews see e.g. [7-9]). Morphologically, most NK cells are large granular lymphocytes (LGL). They are characterized by their intracytoplasmatic azurophilic granules and a high cytoplasm to nucleus ratio. In man, LGL comprise 2%5% of peripheral blood lymphocytes. Early on, NK cells were found in the spleen peritoneal exudate and blood whereas they were scarcely found in lymph nodes, bone marrow, thoracic duct and the thymus. Later, NK cells were also isolated from the liver , tonsils, the epithelial lining of the upper respiratory tract and the lung interstitium [79]. NK cells commonly express certain cell surface markers, defined by monoclonal antibodies against CD 16 (the Fcgammalll receptor for IgG) and NKH-l in humans and NK-l.l/2.1 in mice [7-9]. Additional cell surface markers, some of which define subsets of NK cells, have recently been described [10]. NK cells are of bone marrow origin [6], but their exact lineage is uncertain. They share certain cell surface markers with T cells and also share some characteristics with monocytes. There is now a general belief that mature NK cells are distinct from T lymphocytes [6-10]. The T -cell antigen receptor is not involved in NK cell recognition or cytotoxicity [7- 9] .NK cells do not express CD 3. There is no rearrangement of alpha-, beta-, gamma-, or delta- T cell receptor (TCR) genes and no synthesis of functional TCR messenger ribonucleic acid (mRNA) (even though nonfunctional beta and gamma TCR transcripts may be detected). However, CD3 positive (alpha-beta or gamma-delta) T cells may express, particularly upon activation, MHC nonrestricted cytolytic activity against target cells that are also sensitive to NK cells. According to a new definition proposed at the 5th International Natural Killer Cell Workshop (Hilton Head, S.C., 1988) these cells should not be termed NK cells but rather T cells displaying "NK-like" activity or "nonMHC-requiring" cytolysis [7, 8]. NK cells also respond to various lymphokines and interferons by elevated cytotoxic activity. They have been reported to produce different regulatory lymphokines themselves, e.g. IL-l, IL-2, IL-4, IL-5, interferons and colony stimulating factors. Through such mediators (and maybe yet other unknown factors) NK cells are involved in regulation of haematopoesis and lymphocyte functions [7-9]. Natural resistance to infectious agents may be one of the more important functions for NK cells in vivo [11,12]. Particularly during virus infections, high levels of IFN (primarily alpha or beta), are induced in lymphoid organs. Interferons activate NK cells to a higher level of cytotoxicity and stimulate their blastogenesis and proliferation in vivo. Biron et al. [12] recently described a patient with a complete and persistent absence of NK cells but otherwise normal immune functions. This patient first presented with an overwhelming varicella virus infection requiring treatment with acyclovir and later a life-threatening cytomegalovirus (CMV) infection.
Soon after the discovery of NK cells it became clear that these cells could kill certain tumour cell lines in vitro in spite of the fact that they expressed no or only low amounts of MHC class I molecules [13, 14]. This was a significant difference from cytotoxic T cells, which require the presence of MHC molecules to specifically kill target cells [15]. Further, NK cells were shown to be the effectors in rejection of small numbers of certain transplanted tumour cells [6, 16], in the prevention of metastasis [17] and in bone marrow graft rejection [2, 18]. However, NK cells seem to have no or only little impact on established larger cancers. NK cells are not found within tumours; there is no clonal expansion, but rather a systemic suppression of NK activity [1]. A peculiar rejection mechanism, now attributed to NK cells, is the F I-hybrid resistance [2, 19, 20]. This phenomenon has been instrumental in recognizing the in vivo activity of NK cells and it is of particular importance for the following discussion. Hybrid resistance was first reported in 1958 when Snell [21] observed that homozygous lymphomas grew better in the strain of origin than in F 1 hybrids obtained by crossing this strain with another strain. This F1 hybrid effect violated a principle in tissue transplantation. According to the genetic rules of histocompatibility, an F1 hybrid should accept grafts from either of its parents. The most extensive genetic analysis of the F 1 hybrid effect was carried out by Cudkowicz [in 20] who studied rejection of normal bone marrow cells in F 1 hosts. They explained the phenomenon on the basis of a positive recognition of hypothetical recessive Hh genes expressed in the parental (and on the graft) but suppressed in the F 1 animal [20]. Snell [22] later offered an alternative interpretation in which F 1 hybrid resistance was seen as a result of a combined match and mismatch rather than a complete match between host effector cells and transplanted cells. This theory originated before NK cells were known to be the effector mechanism in hybrid resistance but nevertheless formed the basis for the missing self hypothesis (developed below), providing an alternative explanation to the Hh model. Critical experiments demonstrating that NK cells were the effector mechanism in F1 hybrid resistance were published in 1977 by Kiessling et al. [2]. Before that, Kiessling et al. [23] and Petranyi et al. [24] had reported a correlation between NK activity in vitro and tumour resistance in vivo among F 1 hybrids. Klein et al. [25] extended these conclusions and mapped F 1 hybrid resistance to the H-2 gene complex in several different tumour combinations. Carlson et al. [26] observed a rapid elimination of intravenously injected leukaemia cells whenever these were "mismatched", i.e. H-2K or D products were not present in relation to the host. This elimination occurred in nude mice but not in NK depleted mice. Allogeneic lymphocyte cytotoxicity is a term used for the rapid destruction of intravenously injected allogeneic lym phocytes by unsensitized hosts. Allogeneic lymphocyte cytotoxicity has been reported in several mammalian species and it has been studied most extensively in rats [27]. It is mediated by NK cells and is in certain aspects related to F1 hybrid resistance.
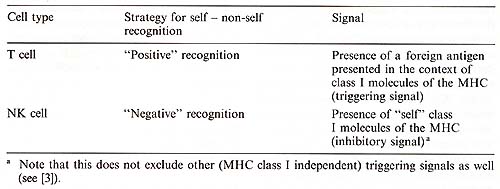
Multicellular organisms need defence systems against destruction of their tissues by foreign invaders as well as against altered endogenous cells. A prerequisite for such a defence reaction is recognition of the potential threat. An organism should be able to discriminate between "self", i.e. everything constituting an integral part of a given individual, and the rest. This recognition could, in theory, be positive or negative [28]. In positive recognition, the organism actively recognizes "non-self" and reacts against it. In negative recognition, there is an active recognition of self and the reaction is triggered only as a consequence of the failure to recognize self. It is well known that higher vertebrates have evolved defence systems based on positive recognition. This is mediated by T and B cells which have clonally distributed receptors generated partly by a random process. The receptor repertoire is then somatically selected for the ability to positively identify "foreignness" either directly (B cells) or in the context of molecules of the MHC (T cells). The missing self hypothesis is based on the second, negative type of recognition. It was suggested that NK cells kill certain targets because they fail to express adequate levels of self MHC class I gene products [3- 5] (Table 1 ). This hypothesis originated from the observations that NK cells mediate rejection of allogeneic lymphoma and bone marrow grafts (H 2a/a rejects H-2b/b) and also, in contrast to cytotoxic T lymphocyte (CTL), F1hybrid anti-parental resistance.
In order to test the missing self hypothesis we chose to work with the wellcharacterized C57Bl/6 derived RBL-5 and EL-4 murine lymphoma cell lines (H2b haplotype). These lymphomas express high levels of H-2 and are highly malignant in the syngeneic host. Our experimental approach was to test the prediction that selection for loss of H-2 class I expression should be accompanied by an increased sensitivity to natural resistance in vivo and in vitro [4, 5]. As a starting point, H-2 deficient variant lines were selected from the RBL-5 and EL-4 lymphomas [5, 29]. Titrated doses of wild type and variant cells were inoculated either subcutaneously [5, 29], intravenously or intraperitoneally [30] in small groups of mice, age matched and usually littermates, in several independent experiments and tumorigeneicity was scored. This strategy minimized the risk that random fluctuations in the quality of the respective cell suspensions would be responsible for differences in the ability to form tumours. In line with the prediction of the hypothesis, the H-2 class I deficient RBL-5 and EL-4 variants were rejected in syngeneic C57BL/6 mice after a small tumour inoculum (1000 to 100.000 cells), whereas mutagenized but non-selected, H-2 positive, wild type lines were highly tumorigenic in the corresponding doses [5, 29]. The H-2 deficient cells required a 1000 to 10.000-fold higher dose than the H-2 positive cells to induce more than 50% tumour take irrespective if the cells were inoculated subcutaneously, intravenously or intraperitoneally [5,29, 30]. The rejection of the H-2 deficient lines showed several characteristics of an NK mediated response, including thymus independence and no requirement for preimmunization [5, 29]. The resistance was weakened (but not totally abrogated) by 400 rad irradiation [29], and it was sufficient to remove asialo-GM1 or NK1.1 positive cells (NK cells) from the animal to abrogate the rejection ([29], unpublished results). Experiments comparing the distribution and survival of isotope prelabeled variant and wild type cells indicated that a rapid elimination of the former took place within 24 h after intravenous injection. These differences in rapid elimination of tumour cells were abolished in NK depleted mice [29]. The above mentioned pattern was observed in all organs studied with one exception the brain (discussed in detail in [30-32]). One possible explanation for the differential rejection patterns of the H-2 positive and H-2 deficient lymphoma cell lines in the syngeneic B 6 mice was a difference in an afferent arm of a NK dependent host response [33]. In this scenario the H-2 deficient cells would recruit NK effector cells which would kill H-2 positive and deficient cells equally well without discriminating between them. However, the differential rejection pattern remained when H-2 positive and deficient cells were inoculated into the same animals, whether in different flanks [29] or mixed in the same inoculum [33]. H-2 deficient cells were selectively eliminated even when they were present in a 10-fold excess compared to H-2 positive cells in the same inoculum [33]. These results suggested that the NK dependent response against H-2 deficient cells was selective in an efferent (effector) arm of the response [33]. In recent experiments we have demonstrated that it is possible to restore the tumorigenicity of ß2-microglobulin ß2m) negative EL-4 cell lines by transfection of ß2m (R. Glas et al., to be published). This indicates that ß2m may act as a tumour growth promoting gene when the host defence is dominated by NK cells . The "missing self' hypothesis predicted that it should be possible to obtain NK mediated rejection of a H-2 positive target provided that the host carried one ( or several) extra MH C class I allele( s) in relation to the target. F 1 hybrid resistance and allogeneic lymphocyte rejection were postulated to be examples of this [4,5]. To directly test this concept, still within the RBL-5 model, we used the transgenic strain D 8 generated by Bieberich et al. [34]. The D8 strain was produced by introducing an 8.0-kb genomic fragment containing the H-2Dd gene and 2.5 Kb 5' and 2.0 Kb 3' flanking sequences into B6 zygotes. The transgene product was expressed in different tissues in the same way as the endogenous H-2b haplotype products, without alterations in the expression of the latter. Tumour growth was followed after subcutaneous inoculation of graded doses of RBL-5 lymphoma cells in D 8 and B 6 control mice [35]. The D8 strain was more resistant to subcutaneous challenge of "previously syngeneic" RBL-5 lymphoma cells than B6 controls. The direct role of the H-2Dd gene in this resistance was investigated by the use of (D 8 XB 6)F 1 crosses and (D8XB6)XB6 backcrosses. The latter showed cosegregation with regard to the Dd expression and lymphoma resistance, both of which were inherited in a pattern consistent with control by a single dominant gene [35]. The resistance to RBL-5 (or other B6 derived lymphomas) in the D 8 strain could be abrogated by pretreating mice with anti-asialo GM1 antiserum or anti-NK1.1 mAb, indicating that NK cells were necessary for the rejection [35] Subsequent studies have shown that the elimination of RBL-5 in D8 is a rapid event taking place within 24 h (P. Höglund et al., J Exp Med, in press). In thus entirely resembles the elimination of RBL-5 H-2 deficient variant cells in B6 mice. A similar pattern was seen when the D 8 strain was grafted with B 6 bone marrow. The D8 recipients had acquired an ability to reject bone marrow from C57BL/6 donors but not from D8 donors and this rejection was dependent on the presence of NK cells in the host [18]. Recessive Hh antigens have been mapped to the D region of the H-2 complex, although rejection did not require expression of the dominant D locus product of the graft (reviewed in [20]). Our data do not address the role of putative Hh antigens at the tumour/graft level. However, no transcripts have been detected from the flanking sequences of the Dd gene carried by the construct (a. Jay, unpublished observations). It is therefore concluded that the Dd gene itself is responsible for the resistance at the level of the host. These results are consistent with the predictions of the missing self hypothesis [3- 5]. In conclusion, the results are in line with an NK cell mediated rejection of small tumour or bone marrow grafts providing that the graft lacks (or expresses greatly reduced levels) of at least one MHC class I allele of the host.
Table 2. Influence of experimental protocol
on NK versus T cell mediated rejection 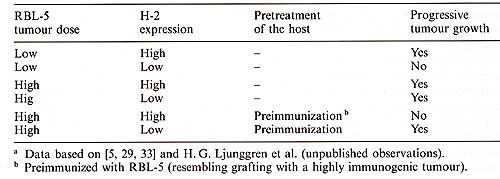
The results obtained with the H-2 deficient lymphoma grafts in vivo predicted that if the critical immunological hosttumour interaction is dominated by NK cells rather than T cells in a given system, up-regulation of MHC class I expression would make tumour cells more malignant because they would survive interactions with NK effectors. This has been observed in studies with different sublines of the mouse B 16 melanoma, pioneered by Taniguchi et al. [37]. H-2 positive melanoma cells gave rise to a high number of metastatic lung colonies, whereas the H-2 low or deficient melanoma cells gave no or only few colonies after intravenous inoculation [37]. Subsequent studies showed that the nonmetastatic H-2 deficient sub lines acquired metastatic capacity if they were pretreated with inteferon (IFN), known to enhance H-2 class I expression, or if the mice were pretreated with anti-asialo OM 1 serum, known to deplete NK cells [38]. In contrast to the result reviewed above, there are several reports in which decreased expression of MHC class I molecules are associated with enhanced tumour growth (reviewed in [39,40]). The apparently conflicting results may depend on the antigenicity of the tumour and/or the experimental protocol used [3, 41]. This can be illustrated with the RBL5 lymphoma, for which opposite results with regard to tumorigenicity were obtained when H-2 positive and deficient variants of this tumour were compared in two different experimental situations (Table 2): 1) A small subcutaneous inoculum in untreated mice led to growth of H-2 positive cells and NK dependent elimination of H-2 deficient cells, and 2) A large subcutaneous inoculum in preimmunized mice resulted in T cell dependent elimination of H-2 positive cells and growth of H-2 deficient cells which overrode the limited non adaptive NK response. Had the tumours only been tested under the latter conditions, we would erroneously have concluded that loss of class I molecules is only associated with enhanced tumorigenicity of the tumour . Thus, there is no obligatory association between reduced H-2 class I expression and increased malignancy. The effect of MHC class I expression on rejection or escape from immunological rejection will depend on the dominant host-tumour interaction. Such interactions may vary in different phases of tumour progression and under different experimental conditions [3, 41]. Tests for the effect of MHC modulation on tumour growth or immunotherapy therefore require careful experimental design to cover the action of different effector mechanisms in vivo. Since T cell responses, once elicited, would playa dominant role in the final outcome of tumour growth, the effect of H-2 changes on the NK defence could rather easily be missed in studies with relatively large tumour grafts and immunogenic tumours. This possibility can be controlled in any system, by using small grafts or short-term assays where rapid rejection of grafted cells is monitored, either in conventional transplantation assays or in tests of the survival of radiolabelled cells [29]. Additional controls can be obtained by the use of mice with either genetically inherited immunodeficiencies ( e.g. nude mice or SCID mice) or mice where different subsets of the host immune defence experimentally has been depleted ( e.g. irradiated, thymectomized, anti-NK1.1 treated, anti-asialo GMl treated, antiL3T4 treated or anti-Ly2 treated mice).
A detailed review of the in vitro NK sensitivity of MHC class I deficient cells has recently been published [3]. A brief summary is given below. The MHC class I deficient RBL-5 and EL-4 murine lymphoma cell lines, used in the in vivo studies described above, showed enhanced NK sensitivity in vitro compared to their wild type counterparts [3, 5, 33]. The association between high NK sensitivity and reduced MHC class I expression in murine models is not confined to lymphoma or melanoma variants derived by mutagenization and selection in vitro. Fibrosarcoma clones with constitutively low H-2 expression derived by cloning without selection were sensitive to NK mediated lysis, while clones (from the same primary tumour) with high levels of H-2 expression were resistant [42]. Increased NK sensitivity of human MHC class I deficient variants was first reported by Harel-Bellan et al. [43] and Storkus et al. [44]. They analysed T -LCL, B-LCL and Band T cell hybrid cell lines. The latter group analysed three different sets of cloned cell lines with corresponding variants that differed in their relative HLA class I expression. While sensitivity correlated with reduced class I expression it did not correlate with class II expression or transferring receptor expression [44]. In one of two more recent confirmatory studies on NK sensitivity of HLA deficient variants [45, 46], IL-2 activated effectors gave the same MHC class I related pattern as fresh NK cells while allospecific CTL lines showed an opposite pattern [45]. The latter study also showed that an intermediate HLA class I expressor (Haplotype loss) variant was moderately NK sensitive (compare to the relatively resistant wild type line ), whereas a weak HLA class I expressor (with an additional down regulation of the remaining haplotype) was highly sensitive [45]. Enhanced NK sensitivity of murine and human MHC class I deficient variant lines does not correlate with a single molecular defect (see [47]). Rather, it appears that increased NK sensitivity can result from different defects in the MHC class I biosynthesis with the common denominator that cell surface expression of class I molecules was impaired. In order to directly study whether MHC class I gene products can affect NK sensitivity it is essential to study cell lines transfected with different genes with the purpose to specifically restore the HLA/H-2 class I expression. The first studies in this direction did not support a role for MHC class I gene products. The murine line 1 carcinoma express little if any H-2Dd in vitro [48]. Transfection of H-2DP into line 1 led to a constitutive and dimethyl sulphoxide (DMSO) inducible expression of H-2DP but this expression had no influence on NK sensitivity. Transfection of H-2Kb into a hepatoma gave a less conclusive result (reduced NK sensitivity in 4/9 experiments, no effect in 5/9 experiments) [49]. The authors further concluded that there was no difference in tumour formation between wild type and transfected cells, and thus that H-2Kb did not affect either NK susceptibility or tumorigenicity. The latter conclusion was based on an i.m. inoculation of 10 high7 cells [49], as opposed to low dose inocula used to study rejection of H-2 deficient cells as reviewed in the present overview (Table 2). However, when Quillet et al. [ 50] transfected the humanß2m gene into theß2m negative human Burkitt lymphoma cell line Daudi, this led to the establishment of a line permanently expressing HLAA10, -All and B-17 class I molecules. This transfected line showed a reduced sensitivity to both NK and lymphokine activated killer (LAK) cell lysis as compared to the HLA class I negative wild type cell line [50]. Transfection of the human HLA class I deficient lymphoblastoid B cell line C 1 R with HLA-A3, HLA-B7 and HLA-Bw58 also led to a reduction in NK sensitivity [51]. Although there was no significant variation among the HLA-A3, -A 7 and -Bw58 alleles, HLA-A2 appeared unable to protect from NK cell lysis [51]. Comparison of amino acid sequence suggests a restricted number of residues which may be relevant to the protective effect. The protection did not extend to H-2DP or Kb transfected C 1 R cells, nor was it seen when IL-2 stimulated NK effector cells were used [51]. Another B cell lymphoblastoid cell line, 0.221, with a selective loss of HLA class I expression was transfected with HLA-Al, HLA-A2, HLAB8, HLA-B5 or an HLA-C gene [46]. Expression ofMHC class I genes reduced NK cell sensitivity, with a general tendency for HLA-B genes to have the most prominent effect. Transfection of human melanoma cells with c-myc is associated with a specific down regulation of class I expression which is most prominent for the B locus products [52]. This is associated with an increased sensitivity to NK cell lysis [53]. Interestingly, this increased NK sensitivity can be overcome by super-transfection of HLA-B7 and B27 genes to the melanoma cells which restores the original NK resistant phenotype (P. Schrier, personal communication). The increased NK sensitivity of melanomas with a cmyc induced HLA- B suppression as well as some of the above-mentioned transfection studies suggested that selective down regulation of certain allelic or locus specific products (and not necessarily all class I molecules) might be sufficient to induce NK sensitivity. This is interesting in relation to two papers describing the failure to induce a significant NK resistance upon transfection with HLA-A2 [54, 55]. There are now two murine analogues to the rescue of HLA in ß2m transfected Daudi cells. Sturmhöfel and Hämmerling [56] selected an H-2 class I deficient line (S 3) from the murine EL-4 cell line. This variant was found to have a defect in ß2m expression and was highly sensitive to NK cell lysis. Transfection of the S 3 line with the ß2m clone restored cell surface H-2 expression and resulted in a considerable decrease in NK sensitivity. Transfection of class II genes had no effect. Blocking of class I molecules with Fab fragments against class I molecules increased NK sensitivity of EL-4 to the level of the S3 variant [56]. The second example is the transfection of the ß2m deficient Y AC-l variant A.H-2- with ß2m. This restored the Y AC-l phenotype with respect to inducible class I expression and a concomitant protection from NK cell lysis after treatment with IFN-gamma [57]. The Y AC-1lymphoma has also recently been transfected with H2Kb under control of the human metallothionein IIA promoter. These transfected H-2Kb positive sublines showed a reduced sensitivity to murine NK cell lysis [58]. At this stage it can be concluded that expression of certain MHC class I molecules can reduce NK sensitivity in several targets. Different molecular models for how MHC class I molecules can protect certain target cells as well as interpretations of the systems where MHC class I genes do not have this effect on NK sensitivity has recently been discussed (see [3]).
In the present overview we have discussed an NK cell defence against MHC class I deficient cells. The data in support of this concept were obtained with murine MHC class I deficient variant cell lines and their corresponding wild type cell lines in in vivo and in vitro studies. Using MHC class I transgenic mice, we have demonstrated that a deficiency of a single host MHC class I allelic product on a tumour cell graft is sufficient to cause rejection. ß2m, one of the three subunits of an MHC class I molecule [59,60], is the first identified molecule that contributes to the IFN-gamma mediated protection from NK cell lysis [57]. This was suggested, although formally not proven, to be mediated through the increased cell surface expression of class I molecules of the MHC. The general concept of a surveillance system geared to detect "missing self' explains some conflicting results regarding the relationship between MHC class I expression, tumorigenicity and metastasis (see [3]). The concept can also explain F l-hybrid resistance and rapid elimination of allogeneic lymphocytes by NK cells. Rapid elimination of allogeneic lymphocytes, transferred during for example pregnancy or sexual contacts where the transferred cells may cause tissue damage by graft versus host or by transmitting infectious agents such as HIV, may be an important defence action by NK cells. From a more general immunological point of view the present results have challenged the notion that discrimination between self and non-self is the only strategy for immunological elimination of aberrant cells in mammals [3-5]. In relation to clinical bone marrow transplantation, the present defence system may be taken into consideration in discussions of host versus graft reactions [18]. The analysis of models where MHC class I molecules clearly do affect NK sensitivity must now focus on detailed molecular events. Experimental strategies for this analysis has recently been described [3]. The mechanism by which an NK cell recognizes a target cell, deficient in self MHC class I expression is unknown. A detailed analysis as to which parts of the MHC class I molecule contribute to the inhibit ion of NK cell lysis must be undertaken. Such an analysis may lead to insights into the recognition event. Other studies must focus on defining receptor-like structures on NK cells. In an "effector inhibition" model [3] certain specific questions could be asked. Does one of the NK receptors resemble a T cell receptor due to a possible MHC class I binding ability? Is such a receptor primed to recognize only presence of self MHC class I molecules, and does it trigger the lytic machinery if appropriate class I molecules are not recognized? It would also be of interest to know when during ontogeny NK cells learn to discriminate between self and non-self. Do NK cells themselves need to express self MHC class I in order to recognize absence of self MHC class I? Are quantitative levels of MHC class Ion the target monitored relative to the MHC class I expression on the NK cell itself? The ultimate model for such and related studies may be ß2m deficient mice as recently described in the literature [61, 62].
1. Whiteside TL, Herberman RB (1989) The role of natural killer cells in human disease. Clin Immunol Immunopathol 53: 1 23 2. Kiessling R, Hochman PS, Hailer 0, Shearer OM, Wigzell H, Cudkowicz a (1977) Evidence for a similar or common mechanism for natural killer cell activity and resistance to hemopoetic grafts. Eur J ImmunoI7:655-663 3. Ljunggren Ha, Kärre K (1990) In search of the missing self: MHC molecules and NK cell recognition. Immunol Today 11:237-244 4. Kärre K (1985) Role of target histocompatibility antigens in regulation of natural killer activity: a reevaluation and a hypothesis. In: Herberman RB, Callewaert D (eds) Mechanisms of cytotoxicity by NK cells. Academic, New York, pp 81-91 5. Kärre K, Ljunggren Ha, Piontek a, Kiessling R (1986) Selective rejection of H-2 deficient lymphoma variants suggests alternative immune defence strategy. Nature 319:675-678 6. Kiessling R, Wigzell H (1979) An analysis of murine NK cells as to structure, function and biological relevance. Immunol Rev 44: 165-208 7. Hercend T, Schmidt RE (1988) Characteristics and uses of natural killer cells. Immunol Today 9:291-293 8. Ades EW, Lopez C (eds) (1989) Natural killer cells and host defence. Karger, Basel 9. Trinchieri a (1989) Biology of natural killer cells. Adv Immunol 47: 187-375 10. Sentman CL, Hacket J, Kumar V, Bennet M (1989) Identification of a subset of murine natural killer cells that mediates rejection of Hh-1 d but not Hh-1 b bone marrow grafts. J Exp Med 170:191-202 11. Welsh RM, Fitzgerald-Bocarsly P (1989) Anti microbial defence by NK cells: Chairmen's summary. In: Ades EW, Lopez C (eds) Natural killer cells and host defence. Karger, Basel, pp 105 -107 12. Biron CA, Byron KS, Sullivan JL (1989) Severe herpesvirus infection in an adolescent without natural killer cells. N Engl J Med 320:1731-1735 13. Trinchieri a, Santoli D (1978) Anti-viral activity induced by culturing lymphocytes with tumor derived or virus-transformed cells. J Exp Med 147:1314-1333 14. Stern P, aidlund M, Örn A, Wigzell H (1980) Natural killer cells mediate lysis of embyonal carcinoma cells lacking MHC. Nature 285:341-342 15. Yedell JW, Bennik JR (1990) The binary logic of antigen processing and presentation to T cells. Cell 62: 203- 206 16. Kärre K, Klein Go, Kiessling R, Klein a, Roder JC (1980) Low natural in vivo resistance to syngeneic leukemias in natural killer-deficient mice. Nature 284:624-626 17. Hanna N, Fidler IJ (1981) Relationship between metastatic potential and resistance to natural killer cell mediated cytotoxicity in three main tumor systems. INCI66:1183-1190 18. öhlen C, Kling a, Höglund P, Hansson M, Scangos a, Bieberich C, Jay a, Kärre K (1989) Prevention of allogeneic bone marrow graft rejection by H -2 transgene in donor mice. Science 246:666-668 19. Cudkowicz a, Bennet M (1971) Peculiar immunobiology of bone marrow allografts. II. Rejection of parental grafts by resistant F 1 hybrid mice. J Exp Med 134:1513-1528 20. Bennet M (1987) Biology and genetics of hybrid resistance. Adv Immunol 41 : 333-445 21. Snell aD (1958) Histocompatibility genes of the mouse. II. Production and analysis of isogenic-resistant lines. I Natl Cancer Inst 21:843877 22. Snell aD (1976) Recognition structures determined by the H-2 complex. Transplant Proc 8: 147 -156 23. Kiessling R, Petranyi a, Klein a, Wigzell H (1976) Non T cell resistance against a mouse Moloney lymphoma. Int J Cancer 17:275-281 24. Petranyi a, Kiessling R, Povey S, Klein a, Hertzenberg L, Wigzell H (1976) The genetic control of natural killer cell activity and its association with in vivo resistance against a Moloney isograft. Immunogenetics 3 : 15- 28 25. Klein Go, Klein a, Kiessling R, Kärre K (1978) H-2 associated control of natural cytotoxcity and hybrid resistance against RBL-5. Immunogenetics 6: 561- 569 26. Carlson aA, Melnychuk D, Meeker MJ (1980) H-2 associated resistance to leukemia transplantation: natural killing in vivo. Int J Cancer 25: 83- 89 27. Heslop BF, Mcneilage LJ (1989) The F1 hybrid effect in allogeneic lymphocyte cytotoxicity. Transplantation 48: 634- 639 28. Klein J (1982) In: Immunology the science of self-nonself discrimination. Wiley, New York 29. Ljunggren HG, Kärre K (1985) Host resistance directed selectively against H-2 deficient lymphoma variants: analysis of the mechanism. J Exp Med 162:1745-1759 30. Ljunggren HG, Yamasaki T, Collins P, Klein G, Kärre K (1988) Selective acceptance of MHC class I deficient tumor grafts in the brain. J Exp Med 167:730-735 31. Yamasaki T, Ljunggren HG, Öhlen C, Klein G, Kärre K (1989) Enhanced H-2 expression and T cell dependent rejection after intracerebral transplantation of the murine lymphoma Y AC-1. Cell Immunol 120:387-395 32. Ljunggren HG, Yamasaki T, Collins VP, Kärre K (1990) Clearence of experimental tumor grafts in the central nervous system. Role of the major histocompatibility complex class I gene products. In: Johansson BB, Owman C, Widner H, (eds) Pathophysiology of the blood brain barrier . Elsevier, Amsterdam, pp 511-522 33. Ljunggren HG, Öhlen C, Höglund P, Yamasaki T, Klein G, Kärre K (1988) Afferent and efferent cellular interactions in natural resistance directed against MHC class I deficient tumor grafts. J Immunol 140:671-678 34. Bieberich C, Yoshioka T, Tanaka K, Jay G, Scangos G ( 1990) Functional expression of a heterologous major histocompatibility complex class I gene in transgenic mice. Mol Cell BioI 7:4003 - 4009 35. Höglund P, Ljunggren HG, Öhlen C, Ährlund-Richter L, Scangos G, Bieberich C, Jay G, Klein G, Kärre K (1988) Natural resistance against lymphoma conveyed by H-2Dd transgene to C57BI mice. J Exp Med 168:1469-1474 36. Höglund P, Glas R, Öhlen C, Ljunggren HG, Kärre K (1991) Alteration of the natural killer repertoir in MHC transgenic mice. J Exp Med 174:327-334 37. Taniguchi K, Kärre K, Klein G (1985) Lung colonisation and metastasis by disseminated B 16 melanoma cells: H -2 associated control at the level of the host and the tumor cell. Int J Cancer 36:503-510 38. Taniguchi K, Petersson M, Höglund P , Kiessling R, Klein G, Kärre K (1987) Interferon gamma induces lung colonization by intravenously inoculated B 16 melanoma cells in parallel with enhanced expression of class I major histocompatibility complex antigens. Proc Natl Acad Sci USA 84: 3405-3409 39. Dotherty PC, Knowles BB, Wettstein PJ (1984) Immunological surveillance of tumors in the context of major histocompatibility complex restriction of T cell function. Adv Cancer Res 42: 1- 65 40. Bernards R (1987) Suppression of MHC gene expression in cancer cells. Trends Genet 3:298301 41. Ljunggren HG, Kärre K (1986) Experimental strategies and interpretation in the analysis of changes in MHC gene expression during tumor progression. J Immunogenet 13:141-151 42. Algarra I, Ohlcn C, Perez M, LJunggren HG, Klein G, Garrido F, Kärre K (1989) NK sensitivity and lung clearance of MHC class I dcficient cells within a heterogeneous fibrosarcoma. Tnt J Cancer44:675-680 43. Harel-Bellan A, Quillet A, Marchiol C, Demars R, Tursz T, Fradelizi D (1986) Natural killer susceptibility ofhuman cells may be regulated by genes in the HLA region on chromosome 6. Proc Natl Acad Sci USA 83: 5688- 5692 44. Storkus WJ, Howell DN, Salter RD, Dawson RD, Cresswell P (1987) NK susceptibility varies inversely with target cell class I HLA antigen expression. J Tmmunol 138:1657-1660 45. Öhlen C, Bejerano MT, Grönberg A, Torsteinsdottir S, Franksson L, Ljunggren HG, Klein E, Klein G, Kärre K (1989) Studies of sublines selected for loss of HLA expression from an EBV transformed lymphoblastoid cell line. J Immunol 142:3336-3341 46. Shimizu Y, Demars R (1989) Demonstration by class I gene transfer that reduced susceptibility of human cells to natural killer cell lysis is inversely correlated with HLA class I antigen expression. Eur J ImmunoI19:447-452 47. Ljunggren HG, Pääbo S, Cochet M, Kling G, Kourilsky P, Kärre K ( 1989) Molecular analysis of H-2-deficient lymphoma cell lines. Distinct defects in biosynthesis and association of MHC class I heavy chains and ß 2-microglobulin observed in cells with increased sensitivity to NK celllysis. J IllmunoI142:2911-2917 48. Dennert G, Landon C, Lord EM, Eahler DW, Frelinger JG (1988) Lysis of lung carcinoma by poly i: c-induced natural killer cells is independent of the expression of class I histocompatibility antigens. J IllmunoI140:2472-2475 49. Nishillura MI, Stroynowski I, Hood L, Ostrand-Rosenberg S (1988) H-2 Kb antigen expression has no effect on natural killer susceptibility and tumorigenicity of a murine hepatoma. J Illmunol 141 : 4403-4409 50. Quillet A, Presse FR, Marchiol-Fournigault C, Harel-Hellan A, Eenbunan M, Ploegh H, Fradelizi D (1988) Increased resistance to non-MHC-restricted cytotoxicity related to HLA A, E expression. Direct demonstration using ß2-llicroglobulin transfected Daudi cells. J Illmunol 141:17-20 51. Storkus WJ, AlexanderJ, PayneJA, Dawson JR, Cresswell P (1989) Reversal of natural killing susceptibility in target cells expressing transfected class I HLA genes. Proc Natl Acad Sci USA 86:2361-2364 52. Versteegh R, Kruse-Wolters K, Plomp AL, van Leeuwen A, Stall NJ, Ploegh HL, Ruiter DJ, Schrier PJ (1989) Suppression of class I human histocompatibility leukocyte antigen by c-llyc is locus specific. J Exp Med 170:621-635 53. Versteegh R, Pletenburg L TC, Plomp AC, Schrier P ( 1989) High expression of the cmyc oncogene renders melanoma cell prone to lysis by natural killer cells. J Illmunol 143 :4331-4337 54. Stall NJ, Kast WM, Voordouw AC, Pastoors EE, van der Hoeven FA, Melief CJM, Ploegh HL (1989) Lack of correlation between levels of MHC class I antigens and susceptibility to lysis of small cellular lung carcinoma (SCLC) by natural killer cells. J Illmunol 142: 4113-4117 55. Leiden JM, Karpinski EA, Gottschalk L, Kornbluth J (1989) Susceptibility to natural killer cell mediated cytolysis is independent of target cell class I HLA expression. J IllmunoI142:2140-2147 56. Sturmhöfel K, Hällmerling GJ (1990) Reconstitution ofH-2 class I expression by gene transfection decreases susceptibility to natural killer cells of an EL-4 class I loss variant. Eur J IllmunoI20:171-177 57. Ljunggren HG, Sturmhöfel K, Wolpert E, Hämmerling GJ, Kärre K (1990) Transfection of ß2-llicroglobulin restores IFN mediated protection from natural killer cell lysis in Y AC-1 lymphoma variants. J IllmunoI145:380-386 58. Carlow DA, Payne U, Hozumi N, Roder JC, Czitroll AA (1990) Class I (H-2 Kb) gene transfection reduces susceptibility of Y AC-1lymphoma targets to natural killer cells. Eur J Illmunol 20:841-846 59. Townsend A, Öhlen C, Eastin J, Ljunggren HG, Foster L, Kärre K (1989) Association of class I major histocompatibility heavy and light chains induced by viral peptides. Nature 340:443-448 60. Ljunggren HG, Stall NJ, Öhlen C, Neefjes JJ, Höglund P, Hellmels MT, Eastin J, Schullacher TNM, Townsend A, Kärre K, Ploegh HL (1990) Empty MHC class 1 molecules come out in the cold. Nature 346:476-480 61. Koller E, Smithies O (1989) Inactivating the ß2-llicroglobulin locus in mouse embryonic stem cells by homologous recombination. Proc Natl Acad Sci USA 86:8932-8935 62. Zijlstra M, Li E, Sajjadi F, Subrallami S, Jaenisch R (1989) Germ-line transmission of a disrupted ß2-llicroglobulin gene produced by homologous recombination in embryonic stem cells. Nature 342:435-438 |