|
* This manuscript is based on a previous publication
[1] and was also presented
In order to understand cancer, it is necessary to identify cancer genes. The search for such genes and for mechanisms that generate such genes must take into consideration that at the cellular level cancer is a very rare event. The kind of cellular transformation that leads to cancer in vivo occurs only in about one out of2 x 10 high 17 mitoses in humans and animalso The basis for this estimate is that most animal and human cancers are derived from single transformed cells and are hence monoclonal [1-5], that humans and corresponding animals represent about 1016 mitoses (assuming 1014 cells that go through an average 102 mitoses), and that about one person in five dies from tumors [6]. The only proven cancer genes are the transforming ( onc ) genes of retroviruses. These are autonomous transforming genes that are sufficient for carcinogenesis [7, 8]. They transform susceptible cells in culture with the same kinetics as they infect tham, and they cause tumors in animals with single-hit kinetics [7,8]. Therefore, these viruses are never associated with healthy animals and are by far the most direct and efficient natural carcinogens. However tumors with retroviruses that contain onc genes are very rare in nature, as only less then 50 cases are recorded from which such viruses were isolated [5, 7-9]. Moreover these viruses have never been reported to cause epidemics of cancer o The probable reasons are that viral onc genes arise naturally only with great difficulty via two or more illegitimate recombinations, and that once arisen they are very unstable because they are not essential for virus replication [7, 8]. Nonessential genes are readily lost due to spontaneous deletion or mutation. Indeed, onc genes were originally discovered by analysis of spontaneous deletions of the src gene, the onc gene of Rous sarcoma virus (RSV) [10,11]. Subsequently, about 20 other viral onc genes were identified in retroviruses [7-9, 12]. All these viral onc genes were originally defined by "transformation-specific" sequences that are dif ferent from the known sequences of essential virus genes [13]. Since onc genes are unstable, they must also be recent additions to retroviruses. Indeed, the cellular genes from which the transformation-specific sequences of oncogenic retroviruses were transduced have been identified in normal cells. This was initially done by liquid hybridization of transformation-specific viral sequences with cellular DNA [14-18], and later by comparing cloned viral onc and corresponding cellular genes [19]. Such cellular genes have since been termed proto-onc genes [7]. The cellular origin of the transformationspecific sequences of retroviral onc genes is frequently presented as a particular surprise [9, 12]. However, cells are the only known source of genetic material from which viruses could transduce genetic information, and viral transduction has been canonical knowledge since phage gamma was first shown by the Lederbergs and Zinder to transduce ßgalactosidase in the 1950s [ 110] .Indeed, viruses are themselves derivatives of cellular genes that have evolved away from their progenitor genes as they acquired their capacity of self-replication.
On the basis of the sequence homology between viral onc genes and proto-onc genes, viral onc genes have been postulated to be transduced cellular cancer genes, and protoonc genes have been postulated to be latent cancer genes or oncogenes [20-29]. According to this view, termed the oncogene concept [29], proto-onc genes are not only converted to transforming genes from without by transducing viruses, but also from within the cell by increased dosage or increased function [20-29]. Activation of latent oncogenes from within the cell is postulated to follow one of five prominent pathways: (a) point mutation [30, 31]; (b) chromosomal translocation that brings the latent oncogene under the control of a heterologous enhancer or promoter [24, 32]; (c) gene amplification [28,29]; (d) activation from a retroviral promoter integrated adjacent to the latent oncogene [9,23-29]; or (e) inactivation of a constitutive suppressor [33]. Thus, this view predicts that latent cancer genes exist in normal cells. However, the existence of latent cancer genes is a paradox, because such genes would be the most undesirable genes for eukaryotic cells. The very essence of enkaryots is cellular cooperativity, rather than autonomy as is typical of cancer cells and prokaryotes. The oncogene concept was a revision of Huebner's oncogene hypothesis, which postulated activation of latent oncogenic viruses instead of latent cellular oncogenes as the cause of cancer [34]. Nevertheless, Huebner's hypothesis remained unconfirmed because most human and animal tumors are virus-negative [9, 12]. Moreover, the retroviruses and DNA viruses that have been isolated from tumors are not directly oncogenic [5], except for the fewer than 50 isolates of animal retroviruses which contain onc genes [8,9, 12]. The oncogene concept was highly attractive at first sight because it derived credibility from the proven oncogenic function of retroviral onc genes, the viral derivatives of proto-onc genes, and because it promised direct access to the long-sought cellular cancer genes in virus-free tumors with previously defined viral onc genes as hybridization probes. Predictably, the hypothesis has focused the search for cellular cancer genes from the 10 high 5 - 10 high 6 genes of eukaryotic cells to the 20 known proto-onc genes [8, 9, 23-29, 43]. The hypothesis makes four testable predictions, namely, (a) that viral onc genes and proto-onc genes are isogenic; (b) that expression of proto-onc genes would cause cancer; (c) that proto-onc genes from tumors would transform diploid cells as do proviral DNAs of viral onc genes; and above all (d) that diploid tumors exist that differ from normal cells only in activated proto-onc genes. Despite record efforts in the past 6 years, none of these predictions has been confirmed. On the contrary, in fact, the genetic and biochemical analyses that have defined essential retroviral genes, viral onc genes, and proto-onc genes during the past 16 years show in reference to (a) that viral onc genes and proto-onc genes are not isogenic [7, 8] (see below). As regards (b), it turned out that most proto-onc genes are frequently expressed in normal cells [8]. Contrary to the expectation in ( c ), none of the 20 known proto-onc genes isolated from tumors functions as a transforming gene when introduced into diploid cells. (The apparent exceptions of proto-ras and proto-myc are discussed below). By comparison, proviral DNAs of retroviral onc genes transform normal cells exactly as the corresponding viruses [9, 12]. And finally, no diploid tumors with activated proto-onc genes, as hypothesized in ( d), have been found except for those caused by viruses with onc genes [35, 36]. Instead of activated oncogenes [8], clonal chromosome abnormalities are a consistent feature of virus-negative tumors [1-4, 37] and also of all those tumors that are infected by retroviruses without onc genes [ 5] .
Harvey proto-ras is the cellular precursor of Harvey, Balb, and Rasheed murine sarcoma viruses, and Kirsten proto-ras is the cellular precursor of the murine Kirsten sarcoma virus [9, 12]. Both proto-ras and the viruses encode a colinear protein, termed p21, of 189 amino acids (Fig.1) [38-44]. In 1982 it was discovered that Harvey proto-ras extracted from a human bladder carcinoma cell line, but not from normal cells, would transform the morphology of a few aneuploid murine cell lines, in particular the NIH 3T3 mouse cell line [30, 31]. Subsequently proto-ras DNAs from some other cell lines and from some primary tumors [8, 38-40] were also found to transform 3T3 cells. Since such proto-ras DNAs behave like dominant and autonomous cancer genes in this morphological assay, they were claimed to be cellular cancer genes [30, 31,43]. The 3T3 cell transforming function of the Harvey proto-ras gene from the bladder carcinoma was reduced to a single point mutation that changed the 12th ras codon of p21 from the normal gly to val [30, 31]. In the meantime, more than 50 different point mutations in five different ras codons have been identified, all of which activate 3T3 cell transforming function [41,42,88]. Since the viral ras genes and proto-ras genes encode the same p21 proteins, whereas most other viral onc genes encode proteins that are dif ferent from those encoded by proto-onc genes (Fig. 1) [7,8], this system has been considered a direct support for the hypothesis that viral onc genes and proto-onc genes are indeed isogenic and hence can become functionally equivalent by point mutations [2631,42-44]. However the following arguments cast doubt or the claims that point mutations are indeed necessary or sufficient to convert proto-ras to a dominant cancer gene: 1. Although most, but not all (see below), proto-ra,s genes with point mutations have been found in tumors or in certain cell lines, ras mutations are very rare in most spontaneous tumors [8, 38-40]. In fact, the glytoval mutation that was originally found in the human bladder carcinoma cell line [30, 31 ] has never been found in a primary tumor [43, 88]. Moreover, even in certain chemically induced or spontaneous tumors in which ras mutations are relatively frequent a consistent correlation between ras mutations and tumors has never been observed [8, 43-45]. Furthermore, it is not known whether in animals the origin of a ras mutation coincides with the origin of the tumor. For example, the ras mutation of the human bladder carcinoma [30, 31] was only found in a cell line 10 years after this line was derived from the original tumor [46]. On the basis of a numerical argument it is also unlikely that point mutations are sufficient to convert proto-ras genes to dominant cancer genes. The frequency of point mutations of eukaryotes is one in 10 high 8 - 10 high 10 nucleotides per mitosis [47, 48]. Thus, about one in 10 high 7 mitoses is expected to generate mutant Harvey ras genes with dominant transforming function, since the diploid human cell contains about 6 x 10 high 9 nucleotides and since 50 different mutations can activate each of two sets of ras genes of diploid cells. By contrast, spontaneous transformation that leads to clonal tumors occurs in fewer than one out of about 2 x 10 high17 mitoses and only a small minority of these contain mutant ras genes. It may be argued, however, that indeed one out of 10 high 7 mitoses generates a tumor cell with activated proto-ras and that the immune system eliminates these cells. However this is unlikely since a point mutation is not an easy target for immunity. Further, animals or humans who are tolerant to ras point mutations would be expected to develop tumors at a very early age, if pointmutated proto-ra,s genes were dominant cancer genes, as the 3T3 assay suggests. Instead, spontaneous human tumors with activated proto-ras are very rare and all were observed in adults [8, 38-40]. Moreover, the argument that cellular oncogenes exist that can be activated by point mutation and then controlled by immunity is hard to reconcile with the existence of a thymic or nude mice which do not develop more spontaneous tumors than other laboratory mice [49]. Furthermore, this view is inconsistent with the evidence that immunosuppressive therapy or thymectomy does not increase the cancer rate of humans [50]. Finally, one would pre dict that in the absence of immunity, as in cell culture, one out of 10 high 7 normal cells should spontaneously transform due to point mutation of Harvey proto-ras alone and probably the same number due to mutation of Kirsten proto-ras [9]. Yet spontaneous transformation of diploid cells in culture is clearly a much less frequent event. In an effort directly to test the hypothesis that ras genes are activated to dominant cancer genes by point mutation, we [41] analyzed whether the transforming function of ras genes does indeed depend on point mutations. Using site-directed mutagenesis we have found that point mutations are not necessary for the transforming function of viral ras genes and of proto-ras genes that had been truncated to be structurally equivalent to viral ras genes [41]. (See also Cichutek and Duesberg this volume. ) 2. Contrary to expectation, the same proto-ras DNAs from human tumors that transform aneuploid 3T3 cells do not transform diploid human [51] or diploid rodent cells [52-54], the initial material of natural tumors. Thus transformation of 3T3 cells does not appear to be a reliable assay for transforming genes of diploid cells. Instead of initiating malignant transformation, mutated proto-ras genes merely alter the morphology and enhance tumorigenicity of aneuploid 3T3 cells. Apparently they activate one of the many morphogenic programs of eukaryotic cells. Observations that untreated 3T3 cells are tumorigenic in nude mice [55-57] are consistent with this view. Thus, proto-ras genes with point mutations are not sufficient to initiate malignant transformation. They only appear as dominant cancer genes in certain aneuploid cells, such as 3T3 cells, based on unknown biochemical effects that alter the morphology of these cells. Furthermore, morphological transformation of 3T3 cells is not ras gene specific. It occurs spontaneously [58] and also upon transfection with several DNA species derived from tumors or tumor cell lines that, like proto-ras, do not transform diploid cells [28,43,44]. Such DNAs are now widely considered as cellular cancer genes [28, 43, 44], although they are not related to viral onc genes and do not transform diploid cells. 3. Assuming that mutated proto-ras genes are cancer genes, like viral onc genes, one would expect diploid tumors that differ from normal cells only in ras point mutation. Contrary to expectation, chromosome abnormalities are consistently found in those tumors in which proto-ras mutations are occasionally found [2, 4]. The human bladder carcinoma cell line, in which the first proto-ra,s mutation was identified, is a convincing example. This cell line contains over 80 chromosomes (instead of 46) and includes rearranged marker chromosomes [46]. In view of such fundamental chromosome alterations, a point mutation seems to be a rather minor event. Indeed among diploid hamster cells transfected with mutated ras genes, only those that developed chromosomal abnormalities upon transfection were tumorigenic [59,60]. Thus, proto-ra,s genes with point mutations are neither sufficient nor proven to be necessary for carcinogenesis and are not autonomous cancer genes as are viral ra,s genes. In addition, there is no kinetic evidence that the origin of the mutation coincides with the origin of the tumors in which it is found. It is consistent with this view that proto-ras mutations that register in the 3T3 cell transformation assay have been observed to occur in vivo in benign hyperplasias, as for example in benign murine hepatomas [61] or in benign, purely diploid mouse skin papillomas that differentiate into normal skin cells [62-66]. Ras mutations have also been observed to arise after carcinogenesis in aneuploid cancer cells [67-69], rather than to coincide with the origin of cancer. By contrast, viral ras genes are sufficient for transformation and thus initiate transformation of diploid cells in vitro and in vivo with single-hit kinetics and concurrent with infection [8,70,71]. This then raises the question as to why viral ras genes are inevitably carcinogenic under conditions under which proto-ras genes with point mutations are not. A sequence comparison between proto-ras genes and the known viral ra,s genes has recently revealed a proto-ra,s-specific exon that was not transduced by any of the known retroviruses with ra,s genes [41]. (See also Cichutek and Duesberg this volume.) It follows, that proto-ra,s and viral ras genes are not isogenic (Fig. 1). Since four different viral ras genes have been shown to lack the same proto-ras exon and since point mutations are not necessary for transforming function, we have proposed that proto-ras genes derive transforming function for diploid cells by truncation of an upstream exon and recombination with a retroviral promoter ([41], see below).
Proto-myc is the cellular precursor of four avian carcinoma viruses, termed MC29, MH2, CMII, and OK10, with directly oncogenic myc genes [8]. The transforming host range of viral myc genes appears to be limited to avian cells, as murine cells are not transformed by cloned proviral DNAs [52, 53, 72]. Nevertheless, it is thought that proto-myc, brought under the control ofheterologous cellular enhancers or promoters by chromosome translocation, is the cause of human Burkitt's lymphoma or mouse plasmacytoma [32, 64, 73]. The following arguments cast doubt on whether such activated proto-myc genes are indeed necessary or sufficient for carcinogenesIs: 1. The human proto-myc gene is located on chromosome 8. This chromosome is typically rearranged in B cell lines derived from Burkitt's lymphomas [8, 32, 64]. However, although chromosome 8 is subjected to translocations, proto-myc is frequently not translocated, and when trans located it is frequently not rearranged [8, 32, 64]. Moreover, no rearrangements of chromosome 8 were observed in about 50% of primary Burkitt's lymphomas; instead, other chromosome abnormalities were recorded [74]. Thus, proto-myc translocation is not necessary for lymphomagenesis. 2. Expression of proto-myc is not consistently enhanced in lymphomas [8]. 3. As yet no proto-myc gene isolated from any tumor has been demonstrated to transform any cells [8]. In an effort to assay transforming function in vivo, a proto-myc gene that was artificially linked to heterologous enhancers was introduced into the germ line of mice [73]. Several of these transgenic mice developed lymphomas after 1-5 months, implying that activated proto-myc had transformed diploid cells. However, the lymphomas of the transgenic mice were all monoclonal [73]. Thus, if the activated proto-myc gene were indeed responsible for the lymphomas, it would be an extremely inefficient carcinogen, because only one of about 10 high 8 "control" B cells of the same mouse [75] with the same transgenic myc gene was transformed. Further, there is no deletion or mutation analysis to show that the activated proto-myc indeed played a direct role in the tumors of the transgenic mice [73]. In contrast, viral myc genes transform all susceptible cells directly and inevitably [8]. 4. If translocated proto-myc were the cause of Burkitt's lymphomas, one would expect all tumors to be diploid and to carry only two abnormal chromosomes, namely, number 8 and the chromosome that was subject to reciprocal translocation with number 8. Instead, primary Burkitt's lymphomas exist with two normal chromosomes 8 that carry other chromosome abnormalities [74]. Thus, translocated protomyc genes are not sufficient or proven to be necessary for carcinogenesis.
It was estimated above that the probability of spontaneous transformation that leads to monoclonal tumors in humans is 2 x 10 high -17 per mitosis. One would expect activation of a preexisting, latent proto-onc gene to be a much more frequent event. For a given proto-onc gene, the probability of activation per mitosis would be the sum of the probabilities associated with each of the putative pathways [28, 29,33] of proto-onc activation. 1. Since the probability of a point mutation per nucleotide per mitosis is about one in 109 [47,48] per diploid cell, the probability that anyone of the 20 known proto-onc genes is activated would be 2 x 20 x 10high -99, assuming only one activating mutation per proto-onc gene. However, it would be 10 high -7 for Harvey-ras alone, since 50 different mutations are thought to activate this gene to a dominant cancer gene (see above). 2. The probability of a given proto-onc gene to be activated by amplification is about one in 10 high 8, considering that about one in 10 high 3-10 high 5 mitoses leads to gene amplification in vitro and possibly in vivo and that about 10 high 3 out of the 10 high 6 kilobases (kb) of eukaryotic DNA are amplified [76, 77]. The probability that anyone of the 20 known proto-onc genes would be activated by amplification would then be 2 x 10 high- 7. 3. The probability of oncogene activation by chromosome translocation depends largely on what distances between a protoonc gene and a heterologous enhancer and which enhancers are considered sufficient for activation. Since distances > 50 kb of DNA have been considered sufficient for activation of proto-myc [9, 64] and proto-abl [9, 78] (the proto-onc gene of murine Abelson leukemia virus [9]), and since an enhancer is likely to be found in every 50 kb of cell DNA, nearly every translocation within a 50-kb radius of a proto-onc gene should be activating. Thus the probability that a given proto-onc gene is activated per translocation would be 5 x 10 high - 5 (50 kb out of 10 high6 kb). The probability that one of the 20 known protoonc genes is activated would then be 10 high - 3 per translocation. Translocation frequencies per mitosis are not readily available. In hamster cells, translocations are estimated to occur with a probability of 10-6 per mitosis [79, 80]. In cells directly derived from mice and humans, even higher frequencies (0.01-0.3) have been observed upon study in vitro [81-83]. The probability of a translocation per meiotic cell division in humans has been determined to be 10 high-3 - 10 high-4, based on chromosome abnormalities in live births [84]. Assuming one translocation in 10 high 4 mitoses, the probability that one out of the 20 known proto-onc genes is activated per mitosis by translocation would then be about 10 high- 7. 4. The probability that a proto-onc gene would be activated from without by the promoter or enhancer of a retrovirus integrated nearby is even higher than those associated with the intrinsic mechanisms. Since retrovirus integration within 1-10 kb of a putative latent cancer gene is considered sufficient for activation [9, 23-29], since retro virus integration is not site-specific [10, 12], and since eukaryotes contain about 10 high6 kb of DNA, a given proto-onc gene would be activated in at least one out of 10 high6 infected cells [5, 8]. The probability that anyone of the 20 known proto-onc genes would be activated would be 2 x 10 high- 5 per infected cell. The sum of these probabilities should reflect the spontaneous transformation frequency of cells per mitosis in vivo and in vi tro. It would be between 10-5 and 10-7. However, it should be at least 10 high - 7 due to Harvey proto-ras mutations alone. Nevertheless, the actual number may be 10 times lower (or about 10 high-8), depending on whether all or only some of these four putative mechanisms could activate a proto-onc gene and depending on whether a given cell is susceptible to transformation by a given onc gene or to a given retrovirus. Instead, spontaneous transformation per mitosis that leads to monoclonal tumors is only about 2 x 10 high-17 in vivo. Thus the expected probability of spontaneous transformation due to activation of preexisting oncogenes differs at least by a factor of 10 high 9 from that observed in diploid cells in vivo. Again it may be argued that spontaneous malignant transformation does indeed occur at the above rates but that immunity eliminates nearly all transformants. However in this case a thymic or nude mice should not exist and the cancer incidence should increase significantly upon immunosuppressive therapy or thymectomy; yet this is not the case [49, 50]. Moreover, diploid cells in culture have not been observed to transform at the above rates. 5. Certain cancers (e.g., retinoblastomas) are thought to be caused by activation of latent oncogenes that are normally suppressed by two allelic suppressor genes [33]. Cancers caused by such genes would be the product of inactivations of two allelic suppressors and thus very rare [33]. In individuals with genetic defects in one putative suppressor allele tumors such as retinoblastomas should occur due to inactivation of the second suppressor allele with the same frequencies as those estimated above for point mutation, translocation, and retrovirus insertion [33]. However, in over 80% of retinoblastomas that occur in individuals without prior genetic defect the putative suppressor genes appear to be normal as judged by chromosome analysis [33], suggesting that other suppressors inhibit the putative retinoblastoma oncogene or that it does not exist. Instead, other chromosomal abnormalities that are always seen in such tumors [33] may be relevant to carcinogenesis (see below). Further, this activation hypothesis predicts that normal cellular DNA would cure retinoblastoma cells upon experimental transfection. Yet this has not been reported. Likewise, it would be expected that experimental, human-nonhuman heterokaryons that have lost chromosomes with suppressor genes would be transformed. It would also be expected that retinoblasts or other cells from individuals with a genetic defect in one suppressor allele would spontaneously transform with the probability of chromosome nondysjunction. Dysjunction has been observed to occur upon cultivation of biopsied murine [85] and human cells [86] with a probability of one in 10high -3 (monosomies) to one in 10 high -4 ( trisomies) per chromosome and mitosis. However, spontaneous transformations have not been described as occurring at this rate. Thus there is as yet no proof for suppressed cancer genes in normal cells.
Because of the consistent difficulties in demonstrating oncogenic function of protoonc genes, a further revision of the oncogene concept has recently been favored. It proposes that "activated" proto-onc genes, like proto-ras or proto-myc, are not autonomous onc genes like their viral derivatives, but are at least necessary for the kind of carcinogenesis that requires multiple cooperating oncogenes [32, 52, 53, 64, 65, 87, 88]. Thus, activated proto-onc genes are proposed to be functionally different, yet structurally equivalent to viral onc genes. According to this theory, activated proto-onc genes would not be expected to register in transformation assays that detect single-hit carcinogens like viral onc genes [7, 8]. However, the hypothesis fails to provide even a speculative explanation as to why activated proto-onc genes are no longer to be considered functionally equivalent to viral onc genes [8]. Clearly, until the postulated complementary cancer genes are identified, this hypothesis remains unproven [8]. The hypothesis also fails to explain why among certain tumors, such as human carcinomas, individual carcinomas are only distinguishable from each other by the presence or absence of activated, putative oncogenes [8,38-40,42-44]. This implies either (a) that unknown oncogenes that do not register in the 3T3 cell assay would cause the same tumors as the putative oncogenes that do, or (b) that the putative oncogenes are not necessary for these tumors.
Genetic and structural analyses of retroviral genes, viral onc
genes, and proto-onc genes and direct comparisons between them have
shown that viral onc genes and proto-onc genes differ both structurally
and functionally. Therefore, we have proposed that viral onc genes
are indeed new genes that do not preexist in normal cells, rather
than being transduced cellular genes [7, 8, 13, 19] (Fig. 1 ). The
original basis for this proposal was the definition of the transforming
gene of avian carcinoma virus MC29 [89] as a genetic hybrid, rather
than a transduced cellular oncogene [90]. It consists of a promoter
and coding elements (deltagag) from an avian retrovirus linked to
3' coding elements from cellular proto-myc (Fig.1) [90]. Initially
this became evident by comparing the structure and map order of
MC29 with that of the three essential retrovirus genes, namely 5'gag-pol-env
3' (Fig.1) [91,92]. Sequence comparison of the viral deltagagmyc
gene with the chicken proto-myc gene provided direct proof that
only a truncated proto-myc gene was present in MC29. Indeed a complete
5' proto-myc exon was missing from the viral deltagag-myc gene [19].
This was apparently not an accident since the same 5' proto-myc
exon was also missing in the three other myc-containing avian carcinoma
viruses MH2 [93, 94], CMII, and OK10 [8,95]. Thus a viral and a
cellular gene functioned as progenitors or proto-onc 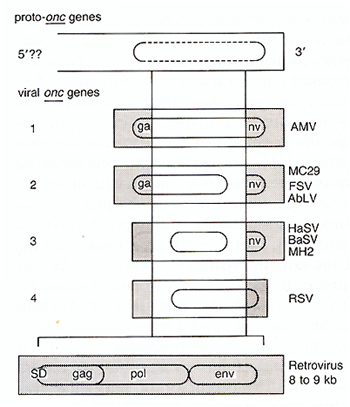
genes of each of the viral recombinant myc genes (Fig. 1 ). More
recently, the four known viral ras genes were each also shown to
lack a 5' proto-ras exon [41] (see above; Fig.1).
The proposal that proto-onc genes derive transforming function by truncation and recombination with retroviral or cellular genes predicts that recombinations among cellular genes could also generate transforming genes. The view that cellular cancer genes are rare recombinants of normal cellular genes is in accord with the fact that rearranged and abnormal chromosomes are the only consistent, transformation-specific markers of tumor cells [2-5,37]. Further, the clonality of chromosome alterations, e.g., the marker chromosomes of tumors [2-5, 37], indicates that tumors are initiated with and possibly caused by such abnormalities as originally proposed by Boveri in 1914 [105]. A major difficulty with the view that spe cific recombination sites among rearranged chromosomes are markers of recombinant cancer genes is that neither the chromosome breakpoints nor the karyotypes of different tumors of the same cell lineage are the same. Although some tumors show typical nonrandom abnormalities, such as the Philadelphia chromosome of chronic myelogenous leukemia and the 8 to 14, 2 and 22 translocations of Burkitt's lymphomas, exceptions are always seen, and the chromosome breakpoints of two different tumors with the same karyotypes are not the same at the nucleotide level [43,74, 106]. Such heterogeneity of breakpoints, and thus of mutation, among otherwise indistinguishable tumors argues either for different transforming genes in the same tumors or against chromosome breakpoints as markers of transforming genes. However, this argument does not take into consideration that together with the microscopic karyotype alterations other submicroscopic mutations may have occurred that could have produced cancer genes. It is consistent with this that tumor cells contain, in addition to microscopic chromosome abnormalities, submicroscopic deletions and restriction enzyme site alterations [107]. Thus, specific marker chromosomes may only be the tip of an iceberg of multiple chromosomal mutations that may have generated cancer genes as well as mutationally activated or inactivated growth control genes. The generation of retroviral onc genes from viral genes and proto-onc genes appears to be a direct model for the process of how cancer genes may be generated by chromosomal rearrangements. Less than 50 isolates of retroviruses with onc genes have been recorded in history [8, 9, 12], although both potential parents of retroviral onc genes are available in many animal or hu man cells because retroviruses are widespread in all vertebrates [5, 9, 12]. This extremely low birth rate of retroviruses with onc genes must then reflect the low probability of generating de novo an oncogenic retrovirus from a proto-onc gene and a retrovirus by truncating and recombining viral and cellular genes via illegitimate recombinations [7, 8, 13]. Clearly, at least two illegitimate recombinations are required (Fig.1): one to link a 3' truncated retrovirus with a 5' truncated proto-onc gene, the other to break and then splice the resulting hybrid onc gene to the 3' part of the retroviral vector. The first of these steps would already generate a "cellular" cancer gene that ought to be sufficient for carcinogenesis. The birth of such a gene would be more probable than that of an oncogenic retrovirus that requires two illegitimate recombinations, but it would be harder to detect than a complete replicating retrovirus with an onc gene. Nevertheless even this would be a rare event. Given that such a recombination would have to take place within the 8-9 kb of a retrovirus (Fig. 1) integrated into the 10 high6-kb genome of a eukaryotic cell and also within an estimated 1-2 kb of a proto-onc gene (Fig. 1), and assuming that translocation or rearrangement occurs with a probability of 10high-4 (see above), the probability of such a recombination per mitosis would be 8 x 10high-6 2x10high-6.10high-4, or 10high-15. That a second illegitimate recombination is required to generate a retrovirus with an onc gene would explain why the occurrence of these viruses is much less frequent than spontaneous transformation due to recombinant cancer genes. This probability may, nevertheless, be higher than the square of 10high-15, since the two events may be linked and since multiple integrated and unintegrated proviruses exist in most infected cells. The probability that illegitimate recombination would generate cancer genes from normal cellular genes would also be very low, since most illegitimate recombination would inactivate genes. The above estimates for the probability of spontaneous trans formation of 2 x 10high-17 per mitosis and of translocation of 10high-4, which would be a minimal estimate for illegitimate recombina tion, suggest that 10 high13 translocations or rearrangements are needed to generate a transforming gene that causes a monoclonal tumor. This could be either a single autonomous transforming gene that is like a viral onc gene or a series of mutually dependent transforming genes [108, 109] that would each arise with a higher probability than an autonomous onc gene. The facts that multiple chromosome alterations are typically seen in tumors [2-4, 37, 74] and that as yet no DNAs have been isolated from tumors that transform diploid cells with single-hit kinetics suggest that most cellular cancer genes are indeed not autonomous carcinogens like viral onc genes. It is consistent with this view that most cellular genes are also not converted to autonomous cancer genes by retroviral transduction via illegitimate recombination and truncation. Only about 20 cellular genes, the proto-onc genes, have been converted to autonomous viral onc genes, although viral transduction via illegitimate recombination is a random event that does not benefit from sequence homology between retroviruses and cells [7, 8, 13]. Thus viral onc genes have not as yet fingered preexisting cellular cancer genes. N o cellular gene is a structural or functional homolog ofa viral onc gene, but the viral onc genes appear to be models for how cancer genes may arise from normal cellular genes by rare truncation and recombination.
I would like to thank S. A. Aaronson, S. Blam, M. Kraus, M. Pech, K. Robbins, S. Tronick and others from the Laboratory of Cellular and Molecular Biology, National Cancer Institute, Bethesda, Maryland, for critical and amusing discussions and generous support during a sabbatical leave, and B. Witkop, National Institute of Arthritis, Diabetes and Digestive and Kidney Diseases, Bethesda, Maryland, for asking many of the basic questions that I try to answer in this manuscript. I also thank my colleagues H. Rubin for encouragement and K. Cichutek, R.-P. Zhou, D. Goodrich, S. Pfaff, and w. Phares, U niversity of California, Berkeley, California, for inspiring comments and their work. This research has been supported by (OIG) National Cancer Institute Grant CA-39915A-Ol and Council for Tobacco Research Grant 1547 and by a Scholarship-in-Residence of the Fogarty International Center, NIH, Bethesda, Maryland.
1. Duesberg PH ( 1987) Cancer genes: rare re combinants instead of activated oncogenes. Proc Natl Acad Sci USA 84:2117-2124 2. Wolman SR (1983) Karyotypic progression in human tumors. Cancer Metast Rev 2:257 293 3. Rowley JD (1984) Introduction: consistent chromosomal alterations and oncogenes in human tumors. Cancer Surveys 3:355-357 4. Trent JM (1984) Chromosomal alterations in human solid tumors: implications of the stem cell model to cancer cytogenetics. Cancer Surveys 3:393-422 5. Duesberg PH (1987) Retroviruses as carcinogens and pathogens: expectations and reali ty .Cancer Res 47: 1199-1220 6. Silverberg E, Lubera J (1986) Cancer statis tics. CA 36:9-25 7. Duesberg PH (1983) Retroviral transform ing genes in normal cells? Nature 304:219 226 8. Duesberg PH (1985) Activated proto-onc genes: sufficient or necessary for cancer? Science 228:669-677 9. Weiss R, Teich N, Varmus H, Coffin J (eds) (1985) RNA tumor viruses; molecular biology of tumor viruses, 2nd edn. Cold Spring Harbor Press, New York 10. Duesberg PH, Vogt PK (1970) Differences between the ribonucleic acids of transforming and nontransforming avian tumor viruses. Proc Natl Acad Sci USA 67:16731680 11. Martin GS, Duesberg PH (1972) The a-subunit on the RNA of transforming avian tumor viruses. I. Occurrence in different virus strains.II. Spontaneous loss resulting in nontransforming variants. Virology 47:494-497 12. Weiss R, Teich N, Varmus H, Coffin J (eds) (1982) RNA tumor viruses; molecular biology of tumor viruses. Cold Spring Harbor Press, New York 13. Duesberg PH (1979) Transforming genes of retroviruses. Cold Spring Harbor Symp Quant. BioI44:13-27 14. Scolnick EM, Rands F, Williams P, Parks WP (1973) Studies on the nucleic acid sequences of Kirsten sarcoma virus. A model for formation of a mammalian RNA-containing sarcoma virus. J ViroI12:458-463 15. Scolnick EM, Parks WP ( 1974 ) Harvey sarcoma virus. A second murine type C sarcoma virus with rat genetic information. J ViroI13:1211-1219 16. Tsuchida N, Gilden RV, Hatanaka M (1974) Sarcoma-virus-related RNA sequences in normal rat cells. Proc Natl Acad Sci USA 71:4503-4507 17. Frankel AE, Fischinger PJ (1976) Nucleo tide sequences in mouse DNA and RNA specific for Moloney sarcoma virus. Proc Natl Acad Sci USA 73:3705-3709 18. Stehelin D, Varmus HE, Bishop JM, Vogt PK (1976) DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 260:170-173 19. Watson DK, Reddy EP, Duesberg PH, Papas TS (1983) Nucleotide sequence analysis of the chicken c-myc gene reveals homologous and unique regions by comparison with the transforming gene of avian myelocytomatosis virus MC29, deltagag-myc. Proc Natl Acad Sci USA 80:2146-2150 20. Bishop JM, Courtneidge SA, Levinson AD, Oppermann H, Quintrell N, Sheiness DK, Weiss SR, Varmus HE (1979) Origin and function of avian retrovirus transforming genes. Cold Spring Harbor Symposia Quant. BioI 44:919-930 21. Karess RE, Hayward WS, Hanafusa H (1979) Transforming protein encoded by the cellular information of recovered avian sarcoma viruses. Cold Spring Harbor Symposia Quant. BioI 44:765-771 22. Wang L-H, Snyder P, Hanafusa T, Moscovici C, Hanafusa H (1979) Comparative analysis of cellular and viral sequences related to sarcomagenic cell transformation. Cold Spring Harbor Symposia Quant. BioI 44:755-764 23. Bishop JM (1981) Enemies within: the genesis of retrovirus oncogenes. Cell 23:5-6 24. Klein G ( 1981) The role of gene dosage and genetic transposition in carcinogenesis. Na ture294:313-318 25. Bishop JM, Varmus H (1982) Functions and origins of retroviral transforming genes in RNA tumor viruses. In: Weiss R, Teich N, Varmus H, Coffin J (eds) RNA tumor viruses; molecular biology of tumor viruses. Cold Spring Harbor Press, New York, pp 999-1108 26. Bishop JM (1982) Oncogenes. Sci Am 246:80-90 27. Bishop JM (1983) Cellular oncogenes and retroviruses. Annu Rev Biochem 52:301 354 28. Varmus H, Bishop JM (1986) Introduction. Biochemical mechanisms of oncogene activity: proteins encoded by oncogenes. Cancer Surv 5:153-158 29. Weiss RA (1986) The oncogene concept. Cancer Rev 2: 1 -17 30. Tabin CJ, Bradley SM, Bargmann CI, Wein berg RA, Papageorge AG, Scolnick EM, Dhar R, Lowy DR, Chang EH (1982) Mechanism of activation of a human oncogene. Nature 300:143-149 31. Reddy EP, Reynolds RK, Santos E, Barbacid M (1982) A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature 300:149-152 32. Leder P, Battey J, Lenoir G, Moulding C, Murphy W, Potter M, Stewart T, Taub R (1983) Translocations among antibody genes in human cancer. Science 227:765-771 33. Knudson AG Jr (1985) Hereditary cancer, oncogenes, and antioncogenes. Cancer Res 45:1437-1443 34. Huebner RJ, Todaro G (1969) Oncogenes of RNA tumor viruses as determinants of cancer. Proc Natl Acad Sci USA 64:10871094 35. Pitot HC (1978) Fundamentals of oncology. Dekker, New York 36. Klein G, Ohno S, Rosenberg N, Wiener F, Spira J, Baltimore D (1980) Cytogenic studies on Abelson-virus-induced mouse leukemias. Int J Cancer 25:805-811 37. Levan A (1956) Chromosomes in cancer tis sue. Ann NY Acad Sci 63:774-792 38. Feinberg AP, Vogelstein MJ, Droller S, Baylin B, Nelkin BD (1983) Mutation affecting the 12th amino acid of the c-Has-ras oncogene product occurs infrequently in human cancer. Science 220:1175-1177 39. Fujita J, Srivastava S, Kraus M, Rhim JS, Tronick SR, Aaronson SA (1985) Frequency of molecular alterations affecting ras protooncogenes in human urinary tract tumors. Proc Natl Acad Sci USA 82:3849-3853 40. Milici A, Blick M, Murphy E, Gutterman JU (1986) c-K-ras codon 12 GGT -CGT point mutation an infrequent event in human lung cancer. Biochem Biophys Res Commun 140:699-705 41. Cichutek K, Duesberg PH (1986) Harvey ras genes transform without mutant codons, apparently activated by truncation of a 5' exon (exon-1). Proc Natl Acad Sci USA 83:2340-2344 42. Lowy DR, Willumsen BW (1986) The ras gene family. Cancer Surv 5:275-289 43. Marshall C (1985) Human oncogenes. In: Weiss R et al. (eds) RNA tumor viruses; molecular biology of tumor viruses. Cold Spring Harbor Press, New York, pp 487558 44. Barbacid M (1986) Mutagens, oncogenes and cancer. Trends Gen 2:188-192 45. Needleman SW, Kraus MH, Srivastava SK, Levine PH, Aaronson SA (1986) High fre quency of N-ras activation in acute myelogenous leukemia. Blood 67:753-757 46. Hastings RJ, Franks LM (1981) Chromosome pattern, growth in agar and tumorigenicity in nude mice of four human bladder carcinoma cell lines. Int J Cancer 27:15-21 47. Wabl M, Burrows PD, Gabain A von, Steinberg A (1984) Hypermutation at the immunoglobulin heavy chain locus in a pre-B cell line. Proc Natl Acad Sci USA 82:479-482 48. Drake JW (1969) Comparative rates of spontaneous mutation. Nature 221 :1132 49. Sharkey FE, Fogh J (1984) Considerations in the use of nude mice for cancer research. Cancer Metast Rev 3:341-360 50. Kinlen Ll (1982) Immunosuppressive therapy and cancer. Cancer Surv 1 :565-583 51. Sager R, Tanaka K, Lau CC, Ebina Y, Anisowicz A (1983) Resistance of human cells to tumorigenesis induced by cloned transforming genes. Proc Natl Acad Sci USA 80:7601-7605 52. Land H, Parada LF, Weinberg RA (1983) Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304:596-602 53. Land H, Parada LF, Weinberg RA (1983) Cellular oncogenes and multistep carcinogenesis. Science 222:771-778 54. Newbold RF, Overell RW (1983) Fibroblast immortality is a prerequisite for transformation by El c-Ha-ras oncogene. Nature 304:648-651 55. Boone CW (1975) Malignant hemangioendotheliomas produced by subcutaneous inoculation of BALB/3T3 cells attached to glass beads. Science 188:68-70 56. Littlefield lW (1982) NIH/3T3 cell line. Science 218:214-216 57. Greig RG, Koestler TP, Trayner DL, Cor win SP, Miles L, Kline T, Sweet R, Yokoyama S, Poste G ( 1985) Tumorigenic and metastatic properties of "normal" and ras-transfected NIH/3T3 cells. Proc Natl Acad Sci USA 82:3698-3701 58. Rubin H, Chu BM, Arnstein P (1983) Heritable variations in growth potential and morphology within a clone of Balb/3T3 cells and their relation to tumor formation. l Natl Cancer Inst 71 :365-373 59. Spandidos DA, Wilkie NM (1984) In vitro malignant transformation of early passage rodent cells by a single mutated human oncogene. Nature 310:469-475 60. Stenman G, Delorme EO, Lau CC, Sager R (1987) Transfection with plasmid pSV2gptEl induces chromosome rearrangements in CHEF cells. Proc Natl Acad Sci USA 84:184-188 61. Reynolds SH, Stowers SI, Maronpot RR, Anderson MW, Aaronson SA (1986) Detection and identification of activated oncogenes in spontaneously occurring benign and malignant hepatocellular tumors of the B6C3F1 mouse. Proc Natl Acad Sci USA 83:33-37 62. Balmain A, Ramsden M, Bowden GT, Smith I (1984) Activation of the mouse cellular Harvey-ras gene in chemically induced benign skin papillomas. Nature 307:658660 63. Balmain A, Pragnell IB (1983) Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature 304:596-602 64. Klein a, Klein E (1984) Oncogene activation and tumor progression. Carcinogenesis 5:429-435 65. Balmain A (1985) Transforming ras oncogenes and multistage carcinogenesis. Br I Cancer 51:1-7 66. Burns FJ, Vanderlaan M, Snyder E, Albert RE (1978) Induction and progression kinetics of mouse skin papillomas. In: Slaga TI, Sivac A, Boutwell RK ( eds ) Carcinogenesis, vol 2. Mechanisms of tumor promotion and cocarcinogenesis. Raven, New York, pp 9196 67. Albino AP, Le Strange AI, Oliff MI, Furth ME, Old LJ (1984) Transforming ras genes from human melanoma: a manifestation of tumour heterogeneity? Nature 308:69-72 68. Tainsky MA, Cooper oS, Giovanella BC, Vande Woude oF (1984) An activated ras N gene: detected in late but not early passage human teratocarcinoma cells. Science 225:643-645 69. Vousden KH, Marshall CJ (1984) Three different activated ras genes in mouse tumors: evidence for oncogene activation during progression of a mouse lymphoma. EMBO J 3:913-917 70. Aaronson SA, Weaver CA (1971) Characterization of murine sarcoma virus (Kirsten) transformation of mouse and human cells. I Gen ViroI13:245-252 71. Hoelzer-Pierce J, Aaronson SA (1982) BALB- and Harvey-murine sarcoma virus transformation of a novel lymphoid progenitor cell. I Exp Med 156:873-887 72. Rapp UR, Cleveland IL, Fredrickson TN , Holmes KL, Morse III HC, Iansen HW, Patschinsky T, Bister K (1985) Rapid induction of hemopoietic neoplasms in newborn mice by a raf(mil)/myc recombinant murine retrovirus. I Virol 55:23-33 73. Adams IM, Harris A W , Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL (1985) The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318:533-538 74. Biggar RI, Lee EC, Nkrumah FK, WhangPeng J (1981) Direct cytogenetic studies by needle stick aspiration of Burkitt's lymphoma in Ghana, West Africa. J Natl Cancer Inst 67:769-776 75. Sprent I (1977) Migration and life span of lymphocytes. In: Loor F, Roelants GE (eds) Band T cells in immune recognition. Wiley, New York, pp 59-82 76. Stark oR (1986) DNA amplification in drug resistant cells and in tumours. Cancer Surv 5:1-23 77. Schimke R T, Sherwood SW, Hill AB, Johnston RN (1986) Overreplication and recombination of DNA in higher eukaryotes: potential consequences and biological implications. Proc Natl Acad Sci USA 83:21572161 78. Heisterkamp N, Stam K, Groffen I, Klein A De, Grosveld a (1985) Structural organization of the bcr gene and its role in the Phi translocation. Nature 315:758-761 79. Kraemer PM, Ray FA, Brothman AR, Bartholdi MF, Cram LS (1986) Spontaneous immortalization rate of cultured Chinese hamster cells. J Natl Cancer Inst 76:703709 80. Ray FA, Bartholdi MF, Kraemer PM, Cram LS (1986) Spontaneous in vitro neoplastic evolution: recurrent chromosome changes of newly immortalized Chinese hamster cells. Cancer Genet Cytogenet 21 :35-51 81. Terzi M, Hawkins TSC (1975) Chromosomal variation and the establishment of somatic cell lines in vitro. Nature 253:361-362 82. Harnden Do, Benn PA, Oxford IM, Taylor AMR, Webb TP (1976) Cytogenetically marked clones in human fibroblasts cultured from normal subjects. Somatic Cell Genet 2:55-62 83. Martin oM, Smith AC, Ketterer Dl, Ogburn CE, Disteche CM ( 1985) Increased chromosomal aberrations in first meta phases of cells isolated from the kidneys of aged mice. Israel J Med Sci 21 :296-301 84. Hook EB (1985) The impact of aneuploidy upon public health: mortality and morbidity associated with human chromosome abnormalities. In: Dellarco VL, Voytek PE, Hoilaender A (eds) Aneuploidy: etiology and mechanisms. Plenum, New York 85. Dzarlieva RT, Fusenig NE (1982) Tumor promoter 12-0-tetradecanoyl-phorbol-13acetate enhances sister chromatid exchanges and numerical and structural chromosome aberrations in primary mouse epidermal cell cultures. Cancer Lett 16:7-17 86. Petersson H, Mitelman F (1985) Nonrandom de novo chromosome aberrations in human lymphocytes and amniotic cells. Hereditas 102:33-38 87. Diamond A, Cooper GM, Ritz J, Lane M-A (1983) Identification and molecular cloning of the human B-Iym transforming gene activated in Burkitt's lymphomas. Nature 305:112-116 88. Varmus H (1984) The molecular genetics of cellular oncogenes. Annu Rev Genet 18:553612 89. Duesberg PH, Bister K, Vogt PK (1977) The RNA of avian acute leukemia virus MC29. Proc Natl Acad Sci USA 74:4320-4324 90. Mellon P, Pawson A, Bister K, Martin GS, Duesberg PH (1978) Specific RNA sequences and gene products of MC29 avian acute leukemia virus. Proc Natl Acad Sci USA 75:5874-5878 91. Wang L-H, Duesberg PH, Beemon K, Vogt PK (1975) Mapping RNase T l-resistant oligonucleotides of avian tumor virus RNAs: sarcoma-specific oligonucleotides are near the poly(A) end and oligonucleotides common to sarcoma and and transformation-defective viruses are at the poly(A) end. J Virol 16:1051-1070 92. Wang L-H (1978) The gene order of avian RNA tumor viruses derived from biochemical analyses of deletion mutants and viral recombinants. Annu Rev Microbiol 32:561592 93. Kan NC, Flordellis CS, Mark GE, Duesberg PH, Papas TS (1984) Nucleotide sequence of avian carcinoma virus MH2: two potential onc genes, one related to avian virus MC29 and the other related to murine sarcoma virus 3611. Proc Natl Acad Sci USA 81:3000-3004 94. Zhou R-P, Kan N, Papas T, Duesberg P (1985) Mutagenesis of avian carcinoma virus MH2: only one of two potential transforming genes (omegagag-myc) transforms fibroblasts. Proc Natl Acad Sci USA 82:6389-6393 95. Hayf1ick J, Seeburg PH, Ohlsson R, PfeiferOhlsson S, watson D, Papas T, Duesberg PH (1985) Nucleotide sequence of two overlapping myc-related genes in avian carcinoma virus OK 10 and their relation to the myc genes of other viruses and the cell. Proc Natl Acad Sci USA 82:2718-2722 96. Duesberg PH, Bister K, Moscovici C (1980) Genetic structure of avian myeloblastosis virus, released from transformed mye loblasts as a defective virus particle. Proc Natl Acad Sci USA 77:5120-5124 97. Lee W-H, Bister K, Pawson A, Robins T, Moscovici C, Duesberg PH (1980) Fujinami sarcoma virus: an avian RNA tumor virus with a unique transforming gene. Proc Natl Acad Sci USA 77:2018-2022 98. Bentley DL, Groudine M (1986) Novel promoter upstream of the human c-myc gene and regulation of c-myc expression in B-cell lymphomas. Mol Cell BioI 6:3481-3489 99. Duesberg P, Vogt PK, Beemon K, Lai M (1974) Avian RNA tumor viruses: mechanism of recombination and complexity of the genome. Quant Bio139:847-857 lOO. Nunn MF, Seeburg PH, Moscovici C, Duesberg PH ( 1983) Tripartite structure of the avian erythroblastosis virus E26 transforming gene. Nature 306:391-395 101. PfaffSL, Zhou R-P, Young JC, Hayf1ick J, Duesberg PH (1985) Defining the borders of the chicken proto-[ps gene, a precursor of Fujinami sarcoma virus. Virology 146:307314 102. van der Hoorn A, Neupert B (1986) The repressor sequence upstream of c-mos acts neither as polyadenylation site nor as transcription termination region. Nucleic Acids Res 14:8771-8782 103. Ikawa S, Hagino- Yamagishi K, Kawai S, Yamamoto T, Toyoshima K (1986) Activation of the cellular src gene by transducing retrovirus. Mol Cell BioI 6:2420-2428 104. Naharro G, Robbins KC, Reddy EP (1984) Gene product of v-fgr onc: Hybrid protein containing a portion of actin and a tyrosinespecific protein kinase. Science 223:63-66 105. Boveri T (1914) Zur Frage der Entstehung maligner Tumoren. Fischer, Jena 106. Klein G (1983) Specific chromosomal trans locations and the genesis of B-cell-derived tumors in mice and men. Cell 32:311-315 107. Dracopoli NC, Houghton AN, Old LJ (1985) Loss of polymorphic restriction fragments in malignant melanoma: implications for tumor heterogeneity. Proc Natl Acad Sci USA 82:1470-1474 108. Rous P (1967) The challenge to man of the neoplastic cell. Science 157:24-28 109. Cairns J (1978) Cancer, science and society. Freeman, San Francisco 110. Zinder NO (1953) Infective heredity in bac teria. Cold Spring Harbor Symp Quant BioI 18:261-269 |