|
|
Hämatol. Bluttransf. Vol 29 |
|
* This lecture was also presented at the "International
Conference on RNA Tumor Viruses in Human Cancer," Denver, Colorado,
United States, 10-14 June, 1984.A portion of this lecture will also
be printed as part of a review in Science, May 10,1985
The main objective of cancer molecular biology is to identify cancer genes. Despite fierce efforts, this objective has still not been met [1-3]. As yet the only known cancer genes are the transforming onc genes of retroviruses. Typically these viruses Initiate and maintain cancers with autonomous transforming genes that are dominant in susceptible cells [5]. The discovery of single gene determinants of cancer ill retroviruses has become a precedent that has infected cancer gene research. It has made retroviral onc genes the favorite models of cellular oncogenes, although the relevance of single-gene models to virus-negative tumors is as yet unknown. Fortunately, onc genes are either detrimental or at least useless to the viability of the virus and thus are not maintained by retroviruses. They are the products of rare, genetic accidents, generated by illegitimate recombinations between retroviruses and cellular genes, termed proto-onc genes. About 20 different proto-onc genes corresponding to 20 different retroviral onc genes are known [5]. At this time the normal function of proto-onc genes has not yet been determined. One of them is structurally related to a growth factor, another is related to a growth factor receptor [6], and two appear to be yeast cell cycle genes [6,7]. It is now widely believed that, upon transcriptional or mutational "activation," proto-onc genes function like viral onc genes. Activation is assumed to be the conversion of a nononcogenic proto-onc gene into a carcinogenic variant. Indeed, mutationally altered or transcriptionally activated proto-onc genes have been found in certain tumors. However, the known mutationally or transcriptionally altered proto-onc genes are structurally different from viral onc genes and have not been shown to be the causes of tumors. Consistent with the single gene models set by retroviral onc genes, it has been proposed, recently, that molecularly defined or cloned DNA species from some tumors are autonomous cancer genes, because these DNAs are capable of transforming the morphology of certain preneoplastic cell lines [4]. Despite the popularity of this view, there is no convincing evidence to date that these DNA species can also transform normal cells in culture or that they are the causes of tumors in animals (see below). Circumstantial evidence suggests that most cancers are not caused by single genes but are the products of multiple genes that have been formally divided into initiation and maintenance genes [ 1-3]. Retroviruses without onc genes (leukemia viruses) and DNA viruses are thought to function either as initiation or as maintenance genes in multigene carcinogenesis because these viruses enhance the cancer risk of infected animals. Recently it has been proposed that activated proto-onc genes playa role in multigene carcinogenesis, rather than being autonomous cancer genes. Here the evidence for the views that activated proto-onc genes are sufficient (one gene-one cancer hypothesis) or at least necessary (multigene-one cancer hypothesis) is reviewed. It is concluded, that there is as yet no adequate functional evidence for oncogenicity and no consistent correlation between any proto-onc alteration and a certain tumor. To date viral onc genes are the only proven examples of"activated" proto-onc genes.
Retroviruses with onc genes are the fastestacting, obligatory carcinogens
known to date. Such viruses have only bccn isolatcd from animals
with ncoplasms, while all other retroviruses and all DNA viruses
with oncogenic potential arc regularly isolated from animals without
neoplasms. This is consistent with single-gene carcinogenesis by
retroviruses with onc genes and possible multigene carcinogenesis
with all other viruses. Indeed, retroviral onc genes are the only
genes known that initiate and maintain cancers per se. That they
are necessary for transformation has been proven genetically with
temperature-sensitive (ts) mutants of Rous (RSV) [8], Kirsten (KiSV)
[9], and Fujinami sarcoma viruses [10, 11]; with avian erythroblastosis
virus [ 12]; and with deletion mutants of these and other retroviruses
[ 13- 19]. The most convincing argument, that they are also sufficient
to initiate and maintain neoplastic transformation, is that all
susceptible cells infected by retroviruses with onc genes become
transformed as soon as they are infected. This high transformation
efficiency virtually excludes selection of preneoplastic cells initiated
by another gene. The structural characteristic of retroviral onc
genes is a specific sequence that is unrelated to the three essential
virion genes gag, pol, and env. This onc-specific sequence of retroviruses
is related to one or several proto-onc genes. Typically the oncspecific
sequence replaces csscntial virion genes and thus renders the virus
replication-defective, or it is added to the essential genes as
in the case of RSV and is readily deleted [5, 13, 14, 20]. Since
onc sequences are parasitic and have no survival value for the virus,
onc genes are readily lost by spontaneous deletion [5, 20]. Therefore,
viruses with onc genes are subject to extinction unless maintained
in laboratories. About 17 of the 20 known viral onc genes are hybrids
of coding regions from proto-onc genes linked to coding regions
from essential retroviral genes [20]. The remaining viral onc genes
consist of coding regions from proto-onc genes linked to retroviral
control elements. The identification of hybrid onc genes provided
the first unambiguous clues that viral onc genes and corresponding
cellular proto-onc genes are dif-ferent, since proto-onc genes are
neither related to nor Iinked in the cell to elements of essential
retrovirus genes [21, 22]. Sequence comparisons of cloned genes
have since confirmed and extended that all proto-onc genes and corresponding
viral onc genes are not isogenic [5, 20]. The known viral onc genes
are subsets of proto-onc genes linked to regulatory and coding elements
of virion genes. In our laboratories we are studying the structural
and functional relationships between viral onc genes and corresponding
proto-onc genes, with particular emphasis on the onc genes of the
following avian carcinoma, sarcoma, and leukemia viruses. The onc
gene of avian carcinoma virus MC29 was the first among viral onc
genes to be diagnosed as a hybrid gene [21, 23] (Fig. I). About
one-half of its information ( 1.5 kb) is derived from the gag gene
of retroviruses; the other half (1.6 kb), termed myc, is derived
from the proto-myc gene [22]. The gene is defined by all O-kilodalton
deltagag-myc protein, termed p 110 [21. 24]. The proto-myc gene
of the chicken has at least three exons. The boundaries of the 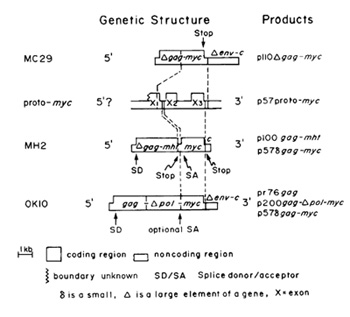
The only clear, although indirect, proof for activation of proto-onc
genes to cancer genes is based on the rare cases in which proto-onc
genes functioned as accidental parents of retroviral onc genes.
It has been deduced from structural analyses of retroviral genes
and proto-onc genes that viral onc genes were generated by transduction
of specific domains from proto-onc genes [5, 20]. Because no significant
sequence homology exists between retroviruses and proto-onc genes,
such transductions must procede via two rare, nonhomologous recombinations
[5,25]5. It is probably for this reason that viral transductions
or ""activations" are extremely rare, even though all cells contain
proto- onc genes and many animal species contain retroviruses without
onc genes. Only 50-100 sporadic cancers from which retroviruses
with onc genes were isolated have been reported and no experimentally
reproducible system of transduction has ever been described [56-58].
Thus, retroviruses with onc genes are the causes of rare, natural
tumors rather than laboratory artifacts. Their role as accidental
progenitors of viral onc genes has made proto-onc genes the focus
of the search for cellular cancer genes. Their possible function
in cancer was initially tested in many laboratories in view of a
""one gene-one cancer" and more recently in view of a ""multigene-one
cancer" hypothesis. The one gene-one cancer hypothesis is similar
to postulates that activation of inactive cellular oncogenes is
sufficient to cause cancer the oncogene hypothesis of Huebner and
Todaro [59]. Some investigators have postulated that activation
is the result of increased dosage of a given proto-onc gene product.
This view, termed the quantitative model, received support from
early experiments which suggested that the src gene of RSV or the
myc gene of MC29 and the corresponding proto-onc genes were equivalents
[60-64]. In the meantime, significant structural and functional
differences between these genes have been found [5,43,44-47, 50]
(see above).
Based on the observation that transcription of the cellular proto-myc
is enhanced in retroviral lymphomas of chicken, it has been postulated
that transcriptional activation of proto-myc is the cause of B-celllymphoma
[64, 74]. Chicken B-cell lymphoma is a clonal cancer that appears
in a small fraction of animals infected by one of the avian leukosis
viruses (which have no onc genes) after latent periods of over 6
months [58]. The hypothesis, termed downstream promotion, postulates
that the gene is activated by the promoter ofa retrovirus integrated
upstream (Fig.3) and that activated proto-myc functions like the
transforming gene of MC29 [64]. Subsequently, samples were found
in which the retrovirus is in tegrated 3' of proto-myc or 5' in
the opposite transcriptional direction. In these cases, the virus
is thought to function like an enhancer of proto-myc [74] (Fig.
3). However, proto-myc differs structurally from the 3-kb delta-gag-myc
gene of MC29 as diagrammed in Figs. land 3 [25, 26]. Further, it
has been argued previously [5] that the hypothesis fails to explain
the origin of about 20% of viral lymphomas in which proto-myc is
not activated [64]; the discrepancies between the phenotype of the
disease and the cancers caused by MC29; the clonality of the tumors,
defined by a single integration site of the retrovirus with regard
to proto-myc; and the long latent period of the disease. Given about
106 kb of chicken DNA and activation of proto-myc by retrovirus
integration within about 5 kb of proto-myc [27,74], one in 2x 105
infections should generate the first tumor cell. Since the chicken
probably has over 107 uncommitted B cells and many more virus particles,
the critical carcinogenic integration event should occur after a
short latent period. The tumor should also not be clonal, since
integration by retroviruses is not site specific and there could
be numerous infections during the latent period of about 6 months.
Further, the model has not been confirmed in murine [75, 76], feline
[35], and bovine [77] leukemia. Instead, the high percentage of
virus-negative fe]ine [35] and bovine [78] lymphomas indicates that
a retrovirus is not even necessary for the disease. Recently, it
was suggested that a mutation, rather than a virus, may have activated
avian proto-myc because mutations have been observed in viral lymphoma
[79]. However, the proto-myc mutations have not been shown to be
the cause of the viral lymphoma. Activation of proto-myc has also
been postulated to cause the retrovirus-negative, human Burkitt's
lymphomas, and mouse plasmacytomas. ]n these cases, chromosome translocation
has been proposed as a mechanism of activating proto-myc function
[70,71,80,8]]. The human proto-myc is related to that of the chicken
from which carcinoma viruses have been derived (Fig.3). The two
genes have unique first exons, similar second exons with unique
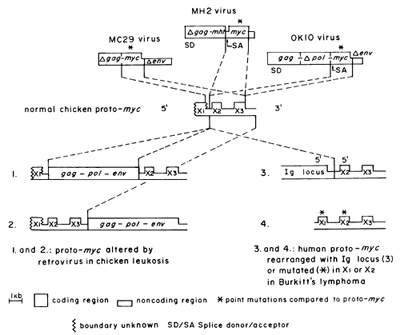
regions, and colinear third exons [25], In man, proto-myc is located
on chromosome 8 and an element of this chromosome is reciprocally
translocated in many Burkitt's lymphoma lines to immunoglobulin
(Ig) loci of chromosome 14 and less frequently of chromosome 2 or
22, Since the crossover points of chromosome 8 are near proto-myc,
translocation was initially suspected to activate proto-myc transcriptionally
by rearranging proto-myc (Fig, I) or by altering its immediate environment
and thus bringing it under the influence of new promoters or enhancers
[80], However, in many lymphomas rearranged proto-myc is not linked
to a new promoter; instead the first presumably noncoding exon is
replaced by the Ig locus, linked to it 5'-5' in the opposite transcriptional
orientation [80] (Fig, 3), Further this model cannot explain how
proto-myc would be activated when the complete proto-myc gene, including
its known promoters and flanking regions, is translocated [70, 72,
82], or recent observations that in a significant minority of Burkitt's
lymphomas proto-myc remains in its original chromosomal location
while a region 3' of proto-myc is translocated [83-87], Despite
these inconsistencies, proto-myc is thought to function as a cellular
oncogene in these tumors. Moreover, there is no consensus at this
time whether proto-myc expression is enhanced in Burkitt's lymphoma
cells, as compared with normal control cells, Some investigators
report elevated expression compared with normal B-Iymphoblasts or
lines [88], while others report essentially normal levels of proto-myc
mRNA [70, 82, 86, 87, 89-92]. Further, en hanced protomyc transcription
is not specific for B-cell lymphomas, since high levels of proto-myc
expression are seen in non-Burkitt's lymphomas [91], in other tumors
[73], and in chemically transformed fibroblast cell lines in which
proto-myc is not translocated or rearranged [43]. The view that
enhanced expression of proto-myc may be sufficient to cause Burkitt's
lymphoma is also challenged by the observations that proto-myc transcription
either reaches cell cycle-dependent peak levels in certain cell
lines [43, 93] or maintains constitutively high levels in embryo
cells similar to those in tumor cells [F. Cuzin (Nice), M. Bywater
(UpPsala), and A. Braithwaite (Canberra), personal communications].
The possibility that mutations of protomyc may correlate with Burkitt's
lymphoma has also been investigated. In some Burkitt's cell lines
mutations have been observed in translocated, but unrearranged,
proto-myc [93, 94] (Fig. 3). Initially it was proposed that these
mutations may activate proto-myc by altering the gene product [94],
but in at least one Burkitt's lymphoma line the coding seq uence
corresponding to proto-myc exons 2 and 3 was identical to that of
the normal gene [82] (Fig. 3). Recently it has been proposed that
mutations in the first noncoding exon may activate the gene [92,
95]. However, there is no functional evidence for this view and
an activating mutation that is characteristic of Burkitt's lymphomas
has not been identified. It is also an open question at this time
whether the first human proto-myc exon is indeed noncoding [82]
or has possibly a large, open reading frame capable of encoding
a major protein [25, 95 a]. A sequence comparison between translocated
proto-myc of a mouse plasmacytoma with the germline proto-myc found
the two genes to be identical except for one nucleotide difference
in the first exon. It was concluded that proto-myc mutations are
not required for oncogenesis [96]. Therefore, no translocation,
rearrangement, elevated expression, or characteristic mutation of
proto-myc is common to all Burkitt's lymphomas investigated. This
casts doubt on the concept that any of the known proto-myc alterations
are a sufficient cause ( or even necessary) for Burkitt's lymphoma.
The question of whether proto-myc has transforming function has
been tested directly using the 3T3 cell transformation assay with
DNA from chicken or human B-celllymphomas. However, no myc-related
DNA was detected even though its presumed functional equivalent,
the delta-gag-myc gene of MC29, is capable of transforming 3T3 cells
[97, 98] and other rodent cell lines [99]. Instead, another DNA
sequence, termed Blym, was identified by the assay [67, 100]. Based
on these results, the role of proto-myc in lymphomas has been interpreted
in terms of a two-gene hypothesis. It has been suggested that activated
proto-myc is necessary but not sufficient to cause the lymphoma
[68, 70]. It is postufated to have a transient early function that
generates a lymphoma maintenance gene, Blym. This gene appears to
be the DNA that transforms 3T3 cells and is thought to maintain
the B-cell tumor. There is no proof for this postulated role of
proto-myc as a lymphoma initiation gene, because the 3T3 cell-transformation
assay does not measure proto-myc initiation function, and because
there is no evidence that the two genes jointly ( or alone) transform
B cells. Furthermore, the hypothesis does not address the question
why proto-myc should have any transforming function at all, if it
is not like MC29. (MC29 does not require a second gene to transform
a susceptible cell.) It is also not known whether Blym is altered
in primary Burkitt's lymphomas, since all of the transfection experiments
were done with DNA from cell lines. It is conceivable that chromosome
translocation involving the proto-myc chromosome 8 may be a specific
but not a necessary consequence, rather than the cause of the lymphoma
[101]. Human B-cell lymphomas with translocations that do not involve
chromosome 8 have indeed been described [ 102, 103]. In the case
of clonal myeloid leukemias with consistent translocations, like
the Philadelphia chromosome, it has been convincingly argued that
translocation is preceded by clonal proliferation of certain stem
cells with the same isoenzyme markers as leukemic cells but without
chromosomal or clinical abnormalities [104]. Perhaps primary Burkitt's
lymphomas should be analysed now and more emphasis should be given
to the question of whether proto-myc alteration contributes to Burkitt's
lymphoma, rather than to speculation about possible mechanisms.
II. Proto-ras Mutations, the Cause of Human and Rodent Carcinomas?
Use of the 3T3 cell assay to measure transforming function of DNA
from a human bladder carcinoma cell line has identified DNA homologous
to the ras gene of Harvey sarcoma virus [66, 105] (Fig.4). Based
on the viral model, the proto-Ha-ras gene is thought to be a potential
cancer gene because it encodes a 21-kilodalton protein, p21, which
is colinear with an onc gene product p21 of Ha-MuSV [106] (Fig.
4). The proto-Ha-ra.s gene from the bladder carcinoma cell line
differs from normal protoHa-ras in a point mutation which alters
the 12th p21 codon in exon I from normal gly to val [66, 107]. This
mutation does not cause overproduction of the ras gene product (p21)
in the 3T3 cell line [66] and does not change known biochemical
properties of p21 [108]. The single-base change is thought to activate
the gene to afunctional equivalent ofHa-MuSV and to be the cause
of the carcinoma because it is the apparent cause for 3T3 cell-transforming
function [66, 109]. However, this mutation has not been found in
over 60 primary human carcinomas, including 10 bladder, 9 colon,
and 10 lung carcinomas [ 110], in 8 other lung carcinomas [III],
and in 14 additional bladder and 9 kidney carcinomas (R. Muschel
and G. Khoury, personal communication). Further, the mutated human
proto-Ha-ras, which transforms 3T3 cells, does not transform primary
rat embryo cells [54, 69] and, more significantly, does not transform
human embryo cell [ 112]. Transformation of primary cells would
be expected from a gene that causes tumors in animals. Thus the
mutated proto-ras gene does not correspond to the viral model which
transforms primary mouse, rat [113,114], 
Fig.4. Comparison of the genetic structures and p21 gene products of the human proto-Ha-ras gene [106,107] and the 5.5-kb RNA genome of Harvey sarcorma virus (Ha-MuSV) [ 132]. HaMuSV is a genetic hybrid of the rat proto-ras gene, a 30S defective retrovirus RNA from rat cells and of Moloney leukemia virus [107,135]
and human cells [115-119]. In addition, the val in the 12th codon of 3T3 cell transforming proto-ras is different for the arg of the viral counterpart [107]. Other mutations have since been found to confer 3T3 cell-transforming function to proto-Ha-ras DNA. Porto-Ha-ras with a mutation in codon 61 was isolated from a human tumor cell line [120]. 3T3 cell-transforming proto-Ha-ras DNAs were also isolated from 2 out of 23 primary urinary tract tumors analyzed. One of these contained a mutation in cod on 61; the other was not identified [121]. The mutations were not found in the normal tissue of the respective patients. Nevertheless, this does not prove that 3T3 cell-transforming function of proto-ras was necessary for tumor formation since each was associated with only lout of 23 histologically indistinguishable tumors. A 3T3 cell-transforming mouse protoHa-ras DNA was also found in some (not all) chemically induced benign papillomas and malignant carcinomas of mice [122]. Since only a small (5%-7%) portion of the benign tumors progressed to carcinomas, it would appear that 3T3 cell-transforming proto-ras was not sufficient to cause the carcinomas, and since not all carcinomas contained the mutation, it would appear that it was not necessary either. A high proportion, i.e., 14 out of 17 methylnitrosourea-induced mammary carcinomas of rats, were found to contain 3T3 celltransforming proto-Ha-ras DNA (M. Barbacid, personal communication). This suggests that the mutation is not necessary for the tumor, although it may be important for tumor progression. The original study reported nine out of nine positives [123]. Moreover, the hormone dependence and high tissue specificity of the carcinogen in this study suggest that other genes must be involved, because mutated proto-ras has been found in association with other tumors and transforms 3T3 cells without hormones. It is plausible that other genes, which may be involved in tumorigenesis but which do not register in the 3T3 assay, were also altered by the carcinogen. In an effort to explain why mutated proto-Ha-ras transforms preneoplastic 3T3 cells, but not rat or human embryo cells, it has recently been proposed that mutated proto-Ha-ras is only one of at least two activated genes that are necessary to induce cancer [54, 55, 69]. This two-gene hypothesis has been tested by transfecting primary rat cells with a mixture of the mutated human proto-Ha-ras and either MC29 provirus or activated proto-myc from mouse plasmacytoma [54], or the EIA gene of adenovirus [69] as helper genes. None of these genes were able to transform rat embryo cells by themselves, but some cells were transformed by the artificial mixed doubles. The study that used the adenovirus virus helper gene showed that proto-rac\' expression varied from high to normal levels in transformed cells and that normal proto-ras was inactive in the assay [69]. The study that used myc-related helper genes did not show that the transformants contained and expressed the added DNAs. It also did not test whether unaltered forms of proto-myc or proto-ras were sufficient for a mixture of these genes to register in this assay. This appears to be a particularly relevant question since a proto-myc clone from a mouse plasmacytoma with an SV40 enhancer at its 3' end but without its natural promoter [71] was reported to be active [54] a]though such a construction is not expected to activate proto-myc. The myc-related genes were proposed to convert rat embryo cells to cells that are capable of dividing indefinitely, like 3T3 cells, a function termed immortalization [54, 55]. The supposed immortalization function of MC29 or of activated proto-myc was not demonstrated independently. The proposal did not explain why an immortalization gene was necessary. Obviously immortalization is necessary to maintain cells in culture. However, immortalization is not necessary for focus formation and probably not for tumor formation since embryo cells are capable of sufficient rounds of mitoses (up to 50) in cell culture and in the animal [ 124]. In the avian system, MC29 transforms primary cells and causes tumors in chicken independently without the benefit of secondary oncogenes, and most MC29 tumor cells are not immortal if tested in cell culture. The failure of maintaining cells from many human tumors in cell culture, under conditions where cells from similar tumors survive, also suggests that immortality may not be an essential criterion of a tumor cell [ 125, 125 a]. There is also no precedent for a function of proto-ras in a multistep transformation mechanism, because the transforming genes of Harvey of Kirsten sarcoma viruses transform rat and mouse embryo cells [113, 114] or human embryo cells [ 115-119] with single-hit kinetics and without helper genes. Moreover, there is no precedent for the artifical mixtures of the two activated proto-onc genes in any natural tumors. Other 3T3 cell-transforming proto-ras genes, namely proto-Ki-ras, which is more closely related to the ras gene of Kirsten sarcoma virus than to Harvey virus, and N-ras, which is related to both viruses, have also been found in tumors or cell lines [ 126]. Proto- Ki-ras encodes a p21 protein that is related to the p21 protein encoded by proto-Ha-ras [107, 126,127]. One group has found 3T3 cell-transforming proto-Ki-ras DNA in three primary human tumors and five tumor cell lines out of 96 samples tested [111,128]. The same group also found 3T3 cell-transforming proto-Ki-ras DNA in one out of eight lung carcinomas tested [III]. The DNA from this tumor, but not that from normal tissue of the same patient, had a mutation in the 12th codon. Obviously the low percentage of 3T3 cellpositives among these tumors raises the question of whether the mutations were necessary for tumorigenesis. In a study of human melanomas, only one of five different metastases from the same human melanoma patient was found to contain 3T3 cell-transforming proto-Kiras DNA [ 129]. A 3T3 cell-transforming Ki-ras DNA was also detected in a metastatic variant but not in a primary methylcholanthrene-induced T-cell lymphoma of mice [130]. An example of a spontaneous protoras mutation appearing in tumor cells cultured in vitro has just been described [ 131 ]. This suggests that these proto-ras mutations were consequences rather than the causes of these tumors. The view that ras mutation is a consequence of tumorigenesis is also consistent with the results that only one ras allele is mutated in some primary tumors [III, 121,127] whereas both alleles are mutated in typical tumor-cell lines [ 110, 111 ]. Since 3T3 cell-transforming or mutated proto-ras genes are only rarely associated with human and murine tumors and since mutated proto-Ha-ras does not transform human or rat embryo cells [54, 69, 112] (proto-Ki-ras was not tested), there is as yet no proof that mutated proto-ras is sufficient or even necessary for any of the above tumors. The failure of the mutated proto-Ha- or Ki-ras to behave like the viral model suggests that structural differences between the cellular and viral genes are responsible (Fig. 4). The 5' end of proto-Ha-ras is not as yet defined [107]. Proto-Ha-ras differs from the 5.5-kb RNA genome of Harvey sarcoma virus [132] in a cell-specific 1-kb DNA region 5' of exon 1 that is preceded by a virus-related region [ 107] and in the sizes (1.2 and 5 kb) of the proto-ras transcripts compared with the genomic viral 5.5-kb mRNA [58, 133, 134]. The cell-specific proto-Ha-ras region is thought to be an intron but it may have another function. The base changes that confer 3T3 celltransforming function to proto-Ha-ras are different from those that set apart viral ras genes from proto-ras [66, 107, 126] (Fig. 4). Proto-Ha-ras with 3T3 cell-transforming function further differs from the viral ra.\" and from normal proto-Ha-ras in point mutations in exons l or 2 [66, 107] (Fig. 4). Moreover, only about 10% of the genomes of Harvey and Kirsten sarcoma viruses are ras related. Each viral RNA contains about 3 kb of genetic information, derived from a rat 30S defective retrovirus RNA [135] which may contribute to the oncogenicity of these viruses (Fig,4). Further, it has been argued that mutated proto-ras is a recessive transforming gene, because both ras alleles are mutated in typical tumor-cell lines, although only one allele is mutated in some primary tumors [ 111, 127]. By contrast, the viral onc gene is dominant. A definitive answer to the question whether ras mutations are dominant or recessive 3T3 cell-transforming genes could be obtained by simultaneous transformation with mutated and normal ras genes. Finally, Ha and Ki- MuSV are not obvious models for proto-ras genes with hypothetical carcinoma function, since these viruses cause predominantly sarcomas.
The preponderance of 3T3 cell-transformation negatives among the above-described tumors suggests that either no genes have caused the negative tumors or that the assay failed to detect them. That only ras-related proto-onc genes have been detected in human tumors signals another limitation of the 3T3 assay. Since the proto-ras mutations found by the 3T3 assay do not transform primary cells, it is possible that they are not relevant for tumor formation. Available data suggest that these are coincidential or consequential rather than cancer causative mutations occurring in tumor cells, because the mutations are not consistently correlated with specific tumors and because in some cases they precede tumor formation and in other they evolve during tumor progression. Despite its effectiveness to transform 3T3 cells, it would follow that mutated proto-ras is not a dominant singular cancer gene, similar to a viral onc gene, and that the test is insufficient to determine whether proto-onc genes cause tumors in animals. The efficiency of the assay to identify cancer genes unrelated to proto-onc genes [4] remains to be determined.
Clearly, proto-onc genes are sometimes mutationally or transcriptionally altered in tumor cells. However, no altered proto-onc gene has been found that looks like a viral onc gene. More importantly, no altered proto-onc gene from tumors investigated functions like a viral onc gene. Altered proto-myc has no transforming function in known assay systems, and altered proto-ras transform 3T3 cells but does not transform rodent or human embryo cells. Thus, altered proto-onc genes are structurally and functionally different from viral onc genes. Moreover, alterd proto-onc genes are not consistently associated with specific tumors. Since there is no functional evidence that altered proto-onc genes transform embryo cells or cause tumors and no consistent correlation between altered proto-onc genes and a specific tumor, the one-gene hypothesis (that altered proto-onc genes are sufficient to cause tumors) is without support. As yet, viral onc genes are the only "activated" proto-onc genes that are sufficient to cause tumors.
The observations that altered proto-onc genes do not behave like viral onc genes and that in some tumors multiple proto-onc genes are altered [73] have been interpreted in terms of a multigene hypothesis. Altered proto-myc has been proposed to cooperate with the Blym gene to cause chicken and human B-cell lymphoma [68]. Altered proto-ras has been proposed to cause carcinomas with other genes, and reported to cooperate in an artificial system with altered proto-myc to transform rat embryo cells in culture [54, 55]. However, there are several reservations about a role of altered proto-myc or proto-ras in multigene carcinogenesis: (a) There is no functional evidence that a combination of altered myc and Blym from lymphomas or that altered ras, together with another gene from carcinomas, transforms appropriate normal test cells. An artificial combination of altered ras in combination with an myc-related or an adenovirus gene was reported to transform primary rat cells. However, it was not reported whether both genes are present and functional in all transformants, and there is no evidence that these artificial ras-helper genes are models for the hypothetical helper genes in tumors with altered ras. (b) The observations that protomyc alterations are not consistently associated with B-cell lymphomas and that proto-ras mutations are only rarely associated with specific carcinomas argue that at least one of two hypothetically synergistic cancer genes is not necessary for these tumors. As yet, no multigene complements that include one or two proto-onc genes have been shown to be consistently associated with specific tumors. ( c) The proposals that altered proto-onc genes play role in a multigene carcinogenesis are a significant departure from the original view that they were equivalents of viral onc genes. The proposals speculate that altered proto-onc genes are necessary but not sufficient for tumor formation and behave like functional subsets of viral onc genes. They do not address the question why these genes are assumed to have unique oncogenic functions that are different from those of the viral models. The ad hoc assumption is without precedent since it is not known whether viral onc genes can be dissociated into complementary or helpergene-dependent genetic subsets. Since there is no functional proof for multiple, synergistic transforming genes and no consistent correlation between at least one altered proto-onc gene and a specific tumor, the view that proto-onc genes are necessary for multigene carcinogenesis is still un proven.
It may be argued that the proto-onc gene alterations that are associated with some cancers playa nonspecific but causative role in carcinogenesis that could be substituted for by another gene. To support this view, it would be necessary to know which other genes could substitute for the role that altered proto-onc genes are thought to play in the origin of cancer. Further, one would have to know whether proto-onc gene alterations are more typical of cancer cells than alterations of other genes and which other genes characteristically undergo alterations in tumor cells. It is likely that unknown events, additional to the known alterations of resident proto-onc genes, are required for the development of cancer [5, 136]. The fact that proto-onc genes share common domains with viral onc genes remains a persuasive argument that proto-onc genes may, under certain conditions, be changed into cancer genes. The evidence that most normal proto-onc genes are expressed in normal cells suggests that cell- specific domains of proto-onc genes may suppress potential oncogenic function. Thus, mutation or removal of suppressors could activate a proto-onc gene, as has been predicted for Burkitt's lymphoma. Clearly, the identification of such suppressors would depend on a complete genetic definition of proto-onc genes. To date, we do not know both termini of any proto- onc gene (except for human proto-myc [79], which is not a protoype of a know oncogenic virus). In addition, virus-specific onc gene elements may also be essential to activate a protoonc gene. In this case, a retrovirus without an onc gene (chronic leukemia virus) could activate a proto-onc gene by a single illegitimate recombination which would form a hybrid onc gene. Such an event would be more probable than the generation of a retrovirus with an onc gene, for which at least two illegitimate recombinations are necessary. DNA technology has made it possible to convert nontransforming DNA from viral or cellular sources to DNA species that transform cell lines or embryo cells. Examples are the proto-mos and proto-ras retroviral L TR recombinants that transform 3T3 cells [49, 50]; the proto-ras, myc, and adenovirus DNA combinations that transform rat embryo cells [51, 66]; or an L TR-mutant proto-ras-SV40 construction that transforms rat embryo cells [137]. Another example is a synthetic gene that consists of a mouse proto-myc gene in which all or part of the first exon is replaced by the L TR of mouse mammary tumor virus. Upon introduction into the germ line, this gene was expressed in II transgenic mice. Only two of these developed mammary tumors after two pregnancies, and not in all mammary glands. It was suggested that the gene may be necessary but not sufficient for the development of these tumors [ 138]. Both the level of expression and the integrity of proto-onc genes were altered in these constructions, since only subsets of proto-onc genes wcrc included. In order to assess the relevance of such iatrogenic transformations to cancer, it would be helpful to determine whether the number of DNA species that can be converted to transforming variants is large or small, and it would be necessary to determine whether any such DNAs ever occur in natural tumors. The most important challenge now is to develop functional assays for cellular cancer genes. Acknowledgements.
1. Rous P (1967) The callenge to man of the neoplastic cell. Science
157.25 la. Berenblum I (1981) Sequential aspects of chemical carcinogenesis.
skin. In. Becker F (ed) Cancer, vol I Plenum, New York |